1Laboratory of Stem Cells and Neuroregeneration, Department of Animal Science, School of Life Sciences, Bharathidasan University, Tiruchirappalli, Tamil Nadu, India.2Faculty Recharge Programme, University Grants Commission (UGC-FRP), New Delhi, India.3Molecular Gerontology Group, Department of Biochemistry, School of Life Sciences, Bharathidhasan University, Tiruchirappalli, Tamil Nadu, India.4Institute of Molecular Regenerative Medicine, Salzburg, Paracelsus Medical University.5Spinal Cord Injury and Tissue Regeneration Center, Salzburg, Paracelsus Medical University, Salzburg, Austria.6Velvio GmbH, Regensburg, Germany.7Austrian Cluster for Tissue Regeneration, Vienna, Austria
通讯作者: Correspondence should be addressed to: Dr. Ludwig Aigner, Institute of Molecular Regenerative Medicine, Paracelsus Medical University, Strubergasse 21, 5020 Salzburg, Austria. Email: ludwig.aigner@pmu.ac.at.Correspondence should be addressed to: Dr. Ludwig Aigner, Institute of Molecular Regenerative Medicine, Paracelsus Medical University, Strubergasse 21, 5020 Salzburg, Austria. Email: ludwig.aigner@pmu.ac.at.
收稿日期:2019-11-19
修回日期: 2020-02-16
接受日期: 2020-02-22
网络出版日期: 2020-07-23
版权声明:
2020 This is an Open Access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
展开
Abstract
Vascular dementia (VaD) is the second leading form of memory loss after Alzheimer's disease (AD). Currently, there is no cure available. The etiology, pathophysiology and clinical manifestations of VaD are extremely heterogeneous, but the impaired cerebral blood flow (CBF) represents a common denominator of VaD. The latter might be the result of atherosclerosis, amyloid angiopathy, microbleeding and micro-strokes, together causing blood-brain barrier (BBB) dysfunction and vessel leakage, collectively originating from the consequence of hypertension, one of the main risk factors for VaD. At the histopathological level, VaD displays abnormal vascular remodeling, endothelial cell death, string vessel formation, pericyte responses, fibrosis, astrogliosis, sclerosis, microglia activation, neuroinflammation, demyelination, white matter lesions, deprivation of synapses and neuronal loss. The transforming growth factor (TGF) β has been identified as one of the key molecular factors involved in the aforementioned various pathological aspects. Thus, targeting TGF-β signaling in the brain might be a promising therapeutic strategy to mitigate vascular pathology and improve cognitive functions in patients with VaD. This review revisits the recent understanding of the role of TGF-β in VaD and associated pathological hallmarks. It further explores the potential to modulate certain aspects of VaD pathology by targeting TGF-β signaling.
KandasamyMahesh, AnusuyadeviMuthuswamy, AignerKiera M, UngerMichael S, KniewallnerKathrin M, SousaDiana M Bessa de, AltendorferBarbara, MrowetzHeike, BogdahnUlrich, AignerLudwig. TGF-β Signaling: A Therapeutic Target to Reinstate Regenerative Plasticity in Vascular Dementia?[J]. Aging and Disease, 2020, 11(4): 828-850 https://doi.org/10.14336/AD.2020.0222
Epidemiology of Vascular Dementia
Dementia describes a set of clinical symptoms affecting cognitive, motor and behavioral aspects, particularly in the geriatric population [1, 2]. Approximately 50 million people suffer from dementia, and its incidence doubles every two decades creating a huge socioeconomic and health care burden worldwide. Vascular Dementia (VaD) contributes to 20-40% of all dementia cases making it the second most common form of dementia after Alzheimer’s disease (AD) [3]. The occurrence of VaD varies among populations. For example, unlike AD, the prevalence of VaD is higher in the Asian compared to the Caucasian population [4]. However, VaD frequently coexists with AD and it is often difficult to distinguish the symptoms between VaD and AD. While AD poses a strong gender bias towards women, the prevalence of VaD is higher in men most likely due to cerebrovascular risk factor being more prevalent in males [5]. Thus far, there are no curative, reparative or regenerative treatments available and therefore, development of therapy for VaD has become an essential but unmet need.
Risk factors of Vascular Dementia
Risk factors for VaD are multifactorial and include aging, illiteracy, genetic predisposition, abnormal conditions and diseases such as hypertension, stroke, diabetes, obesity, coronary infarction, atrial fibrillation, and atypical biochemical blood parameters such as high cholesterol and homocysteine levels. Various lifestyle factors such as smoking, unhealthy diet and physical inactivity have been associated with an increased risk of VaD [2, 6] (Fig. 1). In consequence, reducing or avoiding such risk factors, if possible, is strongly recommended for the prevention of VaD. These include maintenance of healthy blood pressure, prevention or control of diabetes, smoking cessation, maintaining physical fitness and controlling blood cholesterol levels. The rapid increase in cerebrovascular diseases including stroke worldwide suggests that the currently used primary preventive actions against stroke and cardiovascular diseases have very limited efficacy, most likely due to the fact that they function on a voluntary basis with limited self-motivation [7]. This leaves a huge challenge to the scientific community to develop effective therapies to successfully treat VaD.
Figure 1. An overview of Vascular Dementia. The figure summarizes the possible biological and lifestyle risk factors, various forms of VaD, pathological hallmarks of VaD and indicates cognitive deficit as the final outcome of the disease.
Classification of Vascular Dementia
Based on the unstructured diagnostics criteria, VaD has been classified into different types. In 1997, Konno S et al, proposed eight possible forms of VaD, 1) multi-infarct dementia, 2) post-stroke strategic infarcts dementia, 3) multiple subcortical lacunar lesions, 4) Binswanger's disease or arteriosclerotic subcortical leukoence-phalopathy, 5) mixed type of VaD co-occurring with types 1, 2 and 3, 6) haemorrhagic lesion-induced dementia, 7) subcortical vascular dementia, and 8) mixed type of VaD with AD [8, 9]. The aforementioned sub-types of dementias based on vascular dysfunction include several neuropathological syndromes which are all characterized by a reduced cerebral blood flow (CBF) leading to cognitive dysfunctions and memory loss [2]. Some dementias occur after major and/or recurrent strokes in strategically important brain areas and are often considered a major stroke sequel based on large vessel pathology rather than an independent disease [10, 11]. Less common forms of VaD are various types of vasculitis and inherited diseases that affect vessel integrity, such as CADASIL, or the three-prime repair exonuclease (TREX)-1-related thrombotic microvascular disease [12-15]. The predominant form of VaD is based on cerebral small vessel disease, leading to vascular pathologies in deep penetrating arteries and resulting in numerous minor ischemic-hypoxic lesions (multiple lacunes) in deep cortical areas or subcortical areas [16]. Irrespective of the specific subtype, risks include atherosclerosis of cerebral arteries and cerebral amyloid angiopathy [2, 6] (Fig. 1).
Clinical Presentation of Vascular Dementia
In the early phase, VaD can be hardly differentiated from other cognitive deficits such as mild cognitive impairment [6, 17]. As VaD advances, depending on the brain areas that are affected, clinical manifestations of severe cognitive deficits, loss of memory and motor disorders such as gait instability, psychiatric symptoms such as aggressiveness and depression, loss of bladder function, and increasing difficulties with daily routines become clearly evident [1, 2, 6, 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23].
Pathological hallmarks of Vascular Dementia
Irrespective of its origin, the pathogenesis of VaD is quite multifaceted. Upon a closer view, this can involve atherosclerosis, amyloid angiopathy, microbleeding and micro-stroke, blood-brain-barrier (BBB) dysfunction and leakage, infiltration and invasion of blood-derived factors such as circulating immune cells, extracellular vesicles and molecules, abnormal vascular remodeling, endothelial cell death and string vessel formation, pericyte responses, fibrosis, astrogliosis, microglia activation, oxidative stress, neuroinflammation, demyelination and white matter lesions, deprivation of synapses and neuronal loss (Fig. 1) [6, 17-19]. Spontaneous intracerebral hemorrhage can be one of the early pathological features of VaD [24-26]. In the lesioned or degenerative brain, BBB disruption leads to an influx of blood molecules, invasion of blood cells and microbial pathogens into the brain and is associated with inflammatory and immune responses, which can initiate multiple pathways of neurodegeneration [27]. The pathogenic mechanisms rendered by BBB breakdown leads to neuronal injury, synaptic dysfunction, loss of neuronal connectivity and neuronal loss, which have been described in neurodegenerative disorders including VaD [28-31].
The complex pathophysiology in VaD affects neuronal networks involved in behavior, intellectual process, execution and memory, ultimately leading to cognitive deficits [6, 9] (Fig. 1). Considering and accepting this complexity imposes a huge challenge in drug and therapy development for VaD. Most likely, the classical view of a drug with “single mode of action” is inappropriate for a disease like VaD. In consequence, any kind of therapeutic strategy for the diseased brain, including VaD, needs to address a plethora of pathological aspects in a multimodal fashion and needs collectively to include neuroprotection, plasticity, as well as regeneration in the brain. Such an approach most likely needs to be centered around the vascular niche and neurovascular unit, where the various affected cell types meet [32, 33].
Molecular Targets of Vascular Dementia: TGF-β as a potential candidate
On the molecular and genetic level, a few candidates for pathogenesis in VaD have been discussed. For example, polymorphisms and differential expression analyses have identified apolipoprotein E (APOE), methylene-tetra hydrofolate reductase (MTHFR), paraoxonase 1 (PON1), tumor necrosis factor (TNF)-alpha, vascular endothelial growth factor (VEGF), and transforming growth factor (TGF)-β1 as potential candidate factors involved in the pathogenesis of VaD [30, 34]. Among them, TGF-β1 has gained a remarkable scientific interest as it plays a crucial role in angiogenesis, neuroprotection, neuroimmune functions, neural regeneration and synaptic plasticity, which are responsible for cognitive functions in the physiological state [35-38].
TGF-β proteins (TGF-β 1, 2, and 3) are secreted in an inactive form by various cells in various tissues and organs [35]. Thrombospondin (TSP)-1, plasmin, and matrix metalloproteinase (MMP)-2 and -9 trigger the activation process of TGF-β [39]. Besides, the generation of reactive oxygen species (ROS) and the accumulation of an acid milieu in damaged tissues facilitate the activation of TGF-β, thereby leading to the release of TGF-β ligands for the receptor-mediated activation of Smads [40-43]. In particular, TGF-β induces the phosphorylation of Smad2 and Smad3 that bind to Smad4 in the cytoplasm and translocates it into the nucleus [35, 38, 44]. This heteromeric complex acts as a transcription factor to control the expression of various target genes. Thus, TGF-β facilitates transcriptional regulation to create a complex and synergistic higher-order signaling network in various complex biological mechanisms [35, 45] (Fig. 2).
Figure 2. TGF-β signaling in health and disease. Schematic representation of the TGF-β mediated Smad signaling through the TGF-β receptors. TGF-β binds to TGF-β receptor II, which induces the activation of TGF-β receptor I and activates the phosphorylation of Smad2/3 in the cytoplasm. The activated Smad2/3 binds to Smad4 in the cytoplasm and mobilize into the nucleus where they act as a transcription factor. This leads to various cell-type dependent responses essential for the tissue homoeostasis and various physiological functions. Dysregulation of the TGF-β pathway leads to abnormal cellular events and pathological hallmarks that are all part of the VaD pathology.
TGF-β1 regulates cell proliferation, differentiation, maturation and survival of various neuronal and non-neuronal cell types in the brain [38, 46]. Also, it regulates angiogenesis, BBB integrity, neuroinflammation, neuroplasticity and neuronal function [35, 38, 47-50]. The choroid plexus, as well as astrocytes, microglia, and endothelial cells, have been the major sources of TGF-β1 in the brain [51-53]. In the brain, TGF-β1 is typically elevated after acute brain injury and in chronic neurodegenerative pathologies, where it is the main driver of the astroglial scar formation, fibrosis and sclerosis [54-57].
Elevated levels of TGF-β1 in response to cerebro-vascular pathologies, neurodegeneration, neuro-inflammation, and neuroregenerative failure appear to be involved in the progression of dementia [34, 58-62]. Also, genetic variations in TGF-β and TGF-β receptors are considered as the prominent risk factors for VaD. Thus, TGF-β signaling is a therapeutic target candidate in various forms of cognitive disorders. Detailed information on the possible involvement of TGF-β in VaD is limited. In the present review, we focus on TGF-β and its potential role in VaD, in particular, due to its multimodal effects in the brain and neuropathogenesis.
TGF-β and pathogenesis of VaD
Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73].
Strong evidence for the role of TGF-β1 in VaD is derived from studies using mice overexpressing TGF-β1. These mice have originally been generated by Tony Wyss-Coray and colleagues and are characterized by massive deposition of ECM and the formation of hydrocephalus [74]. This mouse model has a higher susceptibility to neuroinflammation triggered by experimental autoimmune encephalitis [75], accelerated disease progression in the context of amyotrophic lateral sclerosis (ALS) [76], severe formation of amyloid plaque pathology in the context of an AD background [69], microvascular degenerations [62], reduced level of neurogenesis [77], and most importantly, learning deficits [78]. More specifically in the context of VaD, these mice show reduced functional vascular reactivity in the sense of a lower dilatation as well as contraction behavior in response to stimuli, capillary degenerations resulting in the formation of string vessels, vascular fibrosis and calcification [79]. Importantly, in the scenario of VaD, the TGF-β1 overexpressing mice show reduced CBF, which correlates with the deposition of amyloid plaques in the blood vessels [80].
Cerebral blood flow and TGF-β in VaD
Reduced CBF is the hallmark of VaD, which may be the result of very diverse conditions such as atherosclerosis, cerebral amyloid angiopathy, microbleeding, micro-strokes and larger territorial stroke. Such damages typically induce a rise in TGF-β1 levels, which locally affects neurodegenerative and neuroregenerative events. The causative role of TGF-β in priming and development of CBF and VaD is obscure. Nevertheless, in patients with hypertension, the main risk factor for VaD, plasma levels of TGF-β1 are markedly increased [81] and this correlates with hypertension-induced tissue and organ damage such as nephrosclerosis and kidney diseases [82]. Thus, the observed changes in CBF seen in neurovascular pathologies might be strongly associated with the increased levels of TGF-mediated defects in the cellular components of the BBB.
The roles of TGF-β in BBB functions
The BBB serves as a remarkable physical and biochemical barricade between the brain and the blood. The BBB is primarily built up by endothelial cells lining the microvessels and sealed through tight junctions. The vessel wall is ensheathed by pericytes and surrounded by end-feet of perivascular astrocytes, microglial cells, and neurons, called neurovascular unit (NVU) [83-87]. In the healthy brain, the BBB protects neurons from blood-derived factors and maintains levels of neurochemical parameters of the internal brain milieu, which are essential for neuroprotection, neuronal functioning and synaptic transmission [84, 88]. This specialized function of the BBB is maintained by the establishment of a mutual trophic interference and three-dimensional vicinity between endothelial cells, pericytes and astrocytes in the brain.
The potential relationship between TGF-β and the BBB is demonstrated in gene knockout studies as complete deletion of TGF-β1, TGF-βR1, TGF-βR2, TGF-βR3 (endoglin) and of Smad4. Those resulted in defects in blood vessel formation and embryonic lethality due to an improper attachment between endothelial and mesothelial cells. Systemic inhibition of TGF-β by soluble endoglin in pregnant rats contributes to the development of Preeclampsia, a disease condition associated with increased BBB leakage in pregnant women. Moreover, a Cre-LoxP based conditional deletion of Smad-4 specifically in brain endothelial cells resulted in BBB breakdown due to disruption in the interaction between pericytes and endothelial cells. Besides, TGF-β contributes to the construction of basement membrane through the secretion of ECM components in endothelial cells and pericytes that are integral parts of the BBB [89-91]. Moreover, the increased autocrine TGF-β signaling in pericytes in diabetic encephalopathy induces basement membrane hypertrophy disrupting the BBB [92]. Taken together, TGF-β plays a key role in the recruitment of pericytes and maintenance of the contact between endothelial cells and pericytes at the BBB (Fig. 3). However, the TGF-β mediated recruitment of astrocytes to the BBB and the complete molecular basis of interactions among cellular components of the BBB remain largely unknown.
In the context of cancer, increased TGF-β facilitates metastasis of tumor cells through the down-regulation of junctional adhesion and tight junction proteins upon the induction of matrix metalloproteases (MMPs) [35, 93]. Similarly, in neuroinflammatory conditions, down-regulation of the expression of junctional adhesion and tight junction proteins in the blood vessel and increased expression of MMPs in association with abnormal TGF-β signaling primes the leakage of BBB, which in turn leads to degeneration of endothelial cells, pericytes drifting and string vessels in the neurovascular systems [94]. Based on the observation from the ablation of the TGF-β signaling pathway in pericytes through retroviral-mediated expression of a truncated C-terminal intracellular kinase domain of TGF-βR2, Sieczkiewicz GJ et al. have described that endothelial cell-specific secretion of TGF-β inhibits the mitotic activities of pericytes through the activation of pericyte contractile protein leading to the BBB breakdown [95].
Figure 3. The intact neurovascular unit and breakdown of the neurovascular unit in VaD. Illustrations of the intact neurovascular unit that typically contains normal blood cells, endothelial cells, basement membrane ensheathed by pericytes and end feet of astrocytes in the brain. The figure also represents the breakdown of the neurovascular unit resulting from the BBB disruption, the crucial pathological hallmarks of VaD such as endothelial cell death and drift of pericyte. The activation of immune cells interaction between the activated microglia and peripheral blood cells leading to the release of TGF-β and breakdown of exosomes and release of macrovesicles in the brain of subjects with VaD.
Abnormal levels of TGF-β and BBB dysfunction in VaD
In a preclinical genetic mouse model of VaD with a point mutation in the NOTCH3 gene, TGF-β signaling is disrupted, and pericyte numbers are reduced leading to BBB breakdown. Notably, endothelial-specific Smad4 deficient animals had increased BBB permeability, BBB breakdown and intracranial haemorrhages [96]. Moreover, the animal models with endothelial cell-specific depletion of TGF-receptors including endoglin have cerebrovascular defects and BBB dysfunctions [97-101]. In humans, mutations in the TGF-β receptors have been linked to hereditary haemorrhagic telangiectasia (HHT). HHT is an autosomal dominant vascular dysplasia, caused by mutations in endoglin and ALK1, which is characterized by dilated vessels and arteriovenous malformations [102]. Eventually, the transgenic model of HHT is characterized by significant structural alterations in cerebral blood vessels, pericyte drift and BBB breakdown [103]. However, the role of TGF-β in the BBB breakdown and vessel leakage in VaD is currently not clear. While many cerebrovascular pathologies, including VaD, have been characterized by the increased level of TGF-β, defects in its downstream pathways and impact of the crosstalk between abnormal levels of TGF-β and other signaling mechanism needs to be considered. Future studies directed towards the potential involvement of TGF-β in BBB dysfunction would signify the therapeutic options for VaD.
The role of TGF-β in vascular remodeling in VaD
Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128].
Recently, endothelial to mesenchymal transition (EndoMT) has been proposed as a potential mechanism involved in VR and BBB breakdown in cerebrovascular pathologies [129, 130]. EndoMT is a cellular process by which endothelial cells dedifferentiate into mesenchymal cells leading to degradation of vascular basement membrane and disintegration of endothelial cells [131]. Moreover, physiological properties and functions of endothelial cells are impaired, and they acquire a migratory phenotype. Notably, cardiac fibrosis, metastatic conditions, systemic sclerosis, multiple sclerosis, stroke, cerebral cavernous malformation, vein stenosis and diabetic retinopathy have been characterized by EndoMT [132-134]. Troletti et al. demonstrated that neuro-inflammation-mediated TGF-β and IL-1 β play a key role in EndoMT in the vasculature of human post-mortem brains with multiple sclerosis and cerebrovascular abnormalities [129]. Moreover, inhibition of TGF-β signaling by neutralizing antibodies, endothelial cell-specific Smad2/3 deletion, shRNA-mediated knockdown of Smad2 and haploinsufficiency of Smad3 minimize EndoMT [135, 136].Therefore, abnormal circulation of TGF-β might contribute to VR and breakdown of BBB through EndoMT in VaD.
Pericyte responses in VaD and the role of TGF-β
Pericytes are multifunctional cells which ensheath the blood vessel and contribute to the regulation of capillary diameter, vasoconstriction and vasodilation, capillary blood flow and extracellular matrix protein secretion [33, 137, 138]. Moreover, pericytes have an immune-modulatory role in the brain [139]. With reference to the origin of the pericytes, both the mesenchymal and neural crest cells have been proposed to give rise to pericytes in many organs. It has recently been reported that pericytes play important roles in the homeostasis of the brain [140-142]. Pericytes are positioned within the BBB between endothelial cells, astrocytes and neurons [138]. Thus, pericytes can integrate and process signals from their adjacent cells to mediate diverse functional responses that are important for brain function in health and disease, including the regulation of the BBB permeability, angiogenesis, clearance of toxic metabolites, phagocytotic activity, capillary hemodynamic responses and neuroinflammation.
In human post-mortem AD brains and transgenic animal models of AD, pericytes are dysfunctional and they undergo degeneration [143-145]. Also, levels of soluble platelet-derived growth factor receptor β (PDGFR-β), a biomarker for pericyte death, are increased in cerebrospinal fluid (CSF) of subjects with mild dementia and in transgenic AD animals [138, 146, 147]. Thus, there is evidence for pericytes being involved in VaD, although the underlying mechanisms are largely unknown. Nevertheless, a familial form of VaD, CADASIL is associated with dysfunction of pericytes in the brain [148].
Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations.
Role of TGF-β on exosomes and microvesicles in VaD
Many physiological and pathophysiological processes are regulated by extracellular vesicles, which are involved in short and large distance intercellular communications independent of direct cell-to-cell contact [160, 161]. Extracellular vesicles contain high amounts of miRNAs and proteins originated in many different cells including peripheral endothelial cells. At present, exosomes and microvesicles have gained huge attention in neurodegenerative diseases including dementia, for example in AD [162-165]. Of note, in the context of AD and VaD, the intermediate products of amyloid-β, such as lower molecular weight amyloid-β oligomers and protofibrils, are transported in extracellular vesicles spreading from neuron to neuron. Those are neurotoxic and act as a source for further aggregation of amyloid-β in the brain [165]. Interestingly, intracellular amyloid-β is often found to be co-localized with exosomes [166, 167]. Remarkably, mobilization of exosomes is regulated by TGF-β. For example, TGF-β is involved in the transport of MMP2 containing exosomes in malignancy disorders [168]. Therefore, it can be speculated that the increased levels of TGF-β in brains with AD and VaD may play a key role in transport and accumulation of amyloid-β responsible for neurodegeneration.
Neuroinflammation and astrogliosis in VaD and the role of TGF-β
Neuroinflammation, like in many other neuro-degenerative diseases, is one of the crucial hallmarks of VaD [146]. It describes a wide range of inflammatory responses within the CNS and is mediated by the production of cytokines, chemokines, and reactive oxygen species. At the cellular level, neuroinflammation involves mainly myeloid cells, i.e. microglia, monocytes, and macrophages, and other peripherally derived immune cells infiltrating the CNS, as well as astrocytes. In response to vascular injuries, microglia are the first cells to respond. Initially classified as M1 pro-inflammatory and anti-regenerative, and M2 anti-inflammatory and pro-regenerative, it is now clear that microglia appear in many different subtypes and phenotypes, with high plasticity and transition between the various types [169-171]. Though microglia have altered activity in VaD even before clinical symptoms and neuropathological hallmarks arise [85, 172], their precise role in VaD is unclear. Nevertheless, under inflammatory conditions, microglia are producers of IL-1b [173], and higher expression of IL-1b has been reported in the brain of hypertensive rats [174]. Also, microglia may contribute to CBF alterations and hypertensive vascular pathology in the brain [175]. Besides microglia, perivascular macrophages might be involved in particular. Monocytes are attracted to the vascular wall by vascular damage, where they differentiate into macrophages, locally secrete matrix proteins, vasculo-protective and pro-angiogenic factors as well as several other growth factors [176], promoting angiogenesis and functional recovery [177].
Neuroinflammation, in particular, microglia activity is strongly regulated by TGF-β. It seems that TGF-β signaling maintains microglia homeostasis. For example, conditional loss of TGF-β signaling in microglia resulted in upregulation of activation and priming markers indicative of a pro-inflammatory stage [178]. Also, TGF-β1 modulates the microglial phenotype and promotes recovery after intracerebral hemorrhage [179]. In the context of VaD, TGF-β regulates microglial activity and induces resistance to hypertension. For example, removal of TGF-β or blocking its signaling before hypertension induction accelerated hypertension progression, and supplementation of TGF-β1 substantially suppressed neuroinflammation and generated immunosuppressive microglia [180]. In summary, TGF-β has, in the context of neuroinflammation and microglia, anti-inflammatory and homeostatic functions (Fig. 4).
Besides monocytes and macrophages, astrocytes are involved in the pathology of VaD. For example, astrogliosis is an acute (7 days) and chronic (3 months) response to artery occlusion in rats [181-183]. Here, in contrast to the effects on microglia, TGF-β seems to have detrimental effects. For example, TGF-β induces astrogliosis and is the main driver of gliotic scarring [74]. In the context of VaD, TGF-β overexpression induces a thickened vascular wall due to accumulation of extracellular matrix proteins [184]. In summary, TGF-β plays a crucial role in neuroinflammation, in particular, in the context of microglial and astroglial cells.
Figure 4. TGF-β and its involvement in neuroinflammation and neurogenesis in the control brain in the context of VaD. Illustration of the regulation of adult neurogenesis by TGF-β at the level of neural stem cell (NSC) self-renewal, transformation of neuroblasts and neuronal differentiation. The intact blood vessel, astrogenesis through astrocyte precursor cell (APS) and oligodendrogenesis from oligodendrocyte precursor cells (OPC) and resident microglial cells are indicated. In the parallel side, the figure indicates the aberrant regulation of adult neurogenesis, activated astrogliosis, microglial activation, accumulation of amyloid β, formation of string vessel, demyelination, neurodegeneration resulting from neuroinflammation mediated abnormal levels of TGF- β in VaD.
Demyelination and white matter lesions in VaD and the role of TGF-β
White matter lesions are defined by white matter hyperintensities in magnetic resonance imaging and are a hallmark of VaD. At the cellular level, white matter lesions are characterized by myelin and oligodendrocyte loss, by axonal damages associated with axonal thinning and the accumulation of varicosities, and by loosening of the axon-oligodendrocyte adhesion [185-188]. Therefore, myelin protection and remyelination are clear targets in VaD. This is, in particular, the case in the aged brain, where remyelination is insufficient [189, 190].
The role of TGF-β in de- and remyelination is still controversially discussed. For example, TGF-β1 is associated with the progression of intracranial deep white matter lesions in humans [191]. In contrast, TGF-β1, which is present in higher levels in the systemic environment, promotes oligodendrocyte maturation, while lower levels in circulating TGF-β prevent remyelination in the spinal cord after toxin-induced demyelination [192]. Also, TGF-β promoted subcortical white matter remyelination, while TGF-β receptor deletion in oligodendroglial progenitors prevents their development into myelinating oligodendrocytes [193].
Loss of synapses and neurodegeneration in VaD and the role of TGF-β
Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia.
A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222].
The regulation of adult hippocampal neurogenesis by TGF-β signaling
Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway.
Regulation of neurogenesis in VaD and the role of TGF-β
In an animal model of VaD, a 2-vessel occlusion (2VO) in experimental rats mimicking chronic cerebral hypoperfusion (CCH) [228, 229], hippocampal neurogenesis is more severely reduced in association with cognitive impairments [230]. Physical exercise rescues the deficits in hippocampal neurogenesis and cognitive functions through the BDNF-CREB pathway in 2VO models [231]. In another study reported by Kwon et al., the inhibition of acetylcholinesterase promotes hippocampal neurogenesis and cognitive function in VaD [232]. Moreover, transplantation of MSCs provided neuroprotection in the hippocampus of 2VO models [233]. Reports on the effects of VaD induced TGF-β levels on adult neurogenesis are limited. As many animal models of AD display a vascular pathology, the regulation of adult neurogenesis by TGF-β can be expected to be similar in VaD as it is described in AD [234, 235]. In corroboration with existing findings on AD and reports generated from experimental models of VaD, it can be presumed that abnormal levels of TGF-β in association with impaired neurogenesis might provide a potential mechanism for vascular abnormalities mediated defects in regenerative plasticity (Fig. 4).
Role of TGF-β signaling in neuroprotection and neuroregeneration in neurodegenerative disorders with dementia
In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology.
Several studies have indicated that the induction of TGF-β during pathogenesis exerts a defensive role in preventing neurodegeneration [219]. Reduced expression of TGF-β and deficient Smad signaling in neurons induces neurodegeneration in the terminal stage of the disease [61]. The reduced neurogenesis seen in the brains of some models of AD could provide a possible explanation for the reduction in TGF-β mediated neurodegeneration in cognitive centers of the pathological brain. Recently, reactive neuroblasts in the diseased brain have been proposed to acquire properties of non-neuronal cells such as microglia, and thus these cells might represent a potential source of TGF-β in pathological brains [223, 253]. The robust activation, proliferation and circulation of microglia, reactive astrocytes and neuroblasts found in early pathogenesis tend to be diminished in the late phases of the diseases [227, 254]. Thus, it can be hypothesized that the overall loss of neuroblasts, non-neuronal and neuronal cells in the brain in the late phase of the disease might lead to the reduced level of TGF-β, thereby fostering neurodegeneration and memory loss.
Figure 5. A proposed mechanism for TGF-β activation in the VaD. The figure represents the proposed hypothesis that interaction between neuroblasts and activated microglial cells might lead to the production of the inactive form of the TGF-β attached with latent transforming growth factor-β binding protein (LTBP) through latency-associated peptide (LAP). The inactive form of TGF-β has generally been stored in the extracellular matrix (ECM). Vascular damage upon mitochondrial defects, release of free radical and acidification of the local microenvironment might be underlying mechanisms of the pathological activation and release of TGF-β in VaD.
A proposed mechanism of TGF-β secretion and activation in VaD
While the potential cellular sources of the increased TGF-β levels are identified, the mechanism for the activation of TGF-β remains unknown in neurodegenerating brains. A possible interaction between dying cells and immune cells is proposed to be involved in the production of TGF-β, which in turn can mediate the inflammatory response and oxidative stress through the generation of inflammatory factors and reactive oxygen species [255, 256] (Fig. 5). Overproduction of free radicals causes oxidative damage to molecular and subcellular components of the cell, thereby leading to cell death in many tissues [257]. Oxidative stress plays a central role in inducing neurodegeneration in many dementia cases [258]. However, the effect of free radical-mediated oxidative stress on the activation of TGF-β has not been clearly established in neurocognitive disorders including VaD. While the increased level of free radicals in the vasculature and reduced bioavailability of vasodilators are associated with endothelial dysfunction leading to vascular dementia, free radicals can lead to acidosis in vascular disorders in association with the degradation of ECM [259]. As mentioned in an earlier chapter, the inactive form of TGF-β can be activated by the free-radical mediated increased acidic condition of the tissue [260]. Thus, the increased level of free radicals might be an underlying cause for triggering the activation of TGF-β in VaD. At the same time, in order to release the active form of TGF-β, the ECM needs to be degraded, which may result in the disruption of the cerebrovascular structure and breakdown of BBB. Thus, free radical-induced activation of TGF-β through the generation of acidic microenvironment could be hypothesized to play a central role in the development of VaD. Recently, Unger MS et al. provided evidence for the association of DCX-expressing T cells and microglia in the vicinity of amyloid plaques in the brain of APP-PS1 transgenic mice and human AD subjects [261]. Moreover, it could be shown that DCX-expressing immune cells exhibit indications of phagocytosis in AD brains [261]. Thus, it can be hypothesized that a potential interaction between the DCX-expressing cells and microglia may be a potential source of TGF-β during neuropathogenesis. Eventually, the elevated TGF-β might be an underlying factor for impaired neurogenesis responsible for memory loss in VaD. Taken together, TGF-β signaling is an ideal therapeutic target to intervene in pathological associated vascular defects, neurodegeneration and promote regenerative plasticity in VaD.
Figure 6. Possible strategies to inactivate TGF-β signaling and expected outcomes in VaD. Illustration of possible ways to inhibit aberrant TGF-β signaling pathways. The activities of TGF-β and TGF-β receptors can be neutralized using various recombinant antibodies or their translation can be blocked using specific set of antisense oligonucleotides. Different synthetic molecules that can inhibit the phosphorylation of the TGF-β RII and Smad2/3 are also indicated. The blockade of pathogenic TGF-β signaling has been expected to mitigate the cognitive deficits through inhibition of various neuropathogenic events and promotion of neuroregenerative plasticity in VaD.
Strategies to modulate TGF-β signaling to mitigate pathogenesis and regenerative potentials in VaD
Town T et al. reported that the inhibition of TGF-β signaling in peripheral immune cells by crossing the CD11c-DNR mice that express the CD11c promoter-driven dominant-negative TGF-β receptor type 2 with Tg2576 mice overexpressing mAPP reduced the brain parenchymal and cerebrovascular β-amyloid deposits and improved the working memory [262]. Therefore, inhibition of overactive TGF-β signaling has been proposed to attenuate neurovascular and neuro-regenerative pathologies in VaD.
TGF-β can be modulated through several distinct approaches (summarized in Figure 6). The main strategies for inhibition of TGF-β include neutralizing antibodies and recombinant fragments that block TGF-β, implementation of compounds and antisense oligonucleotides that interfere with the binding of the ligand to TGF-β receptors, and small molecules that block intracellular phosphorylation of TGF-β receptor 1 and Smad elements. Lerdelimumab (CAT-152) and metelimumab (CAT-192) are recombinant human IgGs generated by phage display technology that block TGF-β1 and TGF-β2 respectively [263]. Recombinant fusion proteins containing the ectodomains of type 2 and type 3 receptors, the targeted pan-TGF-β blocker (TTB) and soluble TGF-β II/Fc have also been used to prevent ligand binding to TGF-β receptors [264]. The synthetic agents such as SB-431542, SB-505124, SD-093, SD-908 and LY2157299 were identified to block the catalytic activity of TGF-βR1 [265]. Moreover, the antisense oligonucleotides generated against the expression of TGF-β or the TGF-β receptors can also be a promising approach to block TGF-β signaling to eventually treat VaD at an early stage of the pathogenesis. However, the safety and efficacy of these agents need to be verified in preclinical and clinical trials. Moreover, inhibition of TGF-β activity may elicit the risk for the anomalies in cell cycle regulation, cytoprotection, immunity and metabolism. Thus, unknown adverse effects of the inhibition of TGF-β related to its normal biological functions and the toxic dosages of the TGF-β agonist may not completely be excluded. Moreover, the overlapping pathophysiology between the VaD and AD needs to be dissected out, for which, the development of a valid experimental model to mimic solely the VaD that reflects the human situation needs to be accomplished. However, further studies are needed to appraise the discussed information and proposed ideas of this article for the most relevant clinical information.
Conclusion
There is ample scientific evidence that strongly supports that aberrant TGF-β signaling is the main underlying mechanism for the pathogenesis of VaD. Increasing evidence has led to TGF-β receiving great scientific attention in recent years because it may play a critical role in the development and progression of neurovascular pathologies and neurocognitive disorders including dementia. Systemic and cerebral alteration of TGF-β in association with the morphological changes of the cerebrovascular system and progressive cognitive disruption may serve as a potential biomarker for VaD. In particular, the multiple aspects of the pathology of VaD where TGF-β might be centrally involved makes the inhibition of TGF-β signaling an attractive and multimodal approach to address the various aspects of VaD. Therefore, the inhibition of aberrant TGF-β might be an ultimate therapeutic strategy for the treatment of VaD subjects.
Acknowledgments
This work was supported by the Austrian Science Funds (FWF) Special Research Program (SFB) F44 (F4413-B23) “Cell Signaling in Chronic CNS Disorders”, by the FWF Project P 31362-B34, and through funding from the European Union’s Seventh Framework Program (FP7/2007-2013) under grant agreements n° HEALTH-F2-2011-278850 (INMiND). MK has been supported by the Faculty Recharge Programme, University Grants Commission (UGC-FRP), New Delhi, India. MK would like to acknowledge a start-up grant from the UGC-FRP, a research grant (SERB-EEQ/2016/000639) and an Early Career Research Award (SERB-ECR/2016/000741) from Science and Engineering Research Board (SERB) under the Department of Science and Technology (DST), Government of India, UGC-SAP and DST-FIST schemes for the support of the infrastructure of the Department of Animal Science, Bharathidasan University.
Global, regional, and national burden of Alzheimer's disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016
XiongXY, LiuL, WangFX, YangYR, HaoJW, WangPF, et al. (2016).
Toll-Like Receptor 4/MyD88-Mediated Signaling of Hepcidin Expression Causing Brain Iron Accumulation, Oxidative Injury, and Cognitive Impairment After Intracerebral Hemorrhage
. Circulation, 134:1025-1038.
[26]
YouS, WangX, LindleyRI, RobinsonT, AndersonCS, CaoY, et al. (2017).
Early Cognitive Impairment after Intracerebral Hemorrhage in the INTERACT1 Study
CaraciF, BattagliaG, BrunoV, BoscoP, CarbonaroV, GiuffridaML, et al. (2011).
TGF-beta1 pathway as a new target for neuroprotection in Alzheimer's disease
. CNS Neurosci Ther, 17:237-249.
[37]
Henrich-NoackP, PrehnJH, KrieglsteinJ (1996).
TGF-beta 1 protects hippocampal neurons against degeneration caused by transient global ischemia. Dose-response relationship and potential neuroprotective mechanisms
. Stroke, 27:1609-1614; discussion 1615.
[38]
KandasamyM, LehnerB, KrausS, SanderPR, MarschallingerJ, RiveraFJ, et al. (2014).
TGF-beta signalling in the adult neurogenic niche promotes stem cell quiescence as well as generation of new neurons
Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain
FloodC, AkinwunmiJ, LagordC, DanielM, BerryM, JackowskiA, et al. (2001).
Transforming growth factor-beta1 in the cerebrospinal fluid of patients with subarachnoid hemorrhage: titers derived from exogenous and endogenous sources
. J Cereb Blood Flow Metab, 21:157-162.
[53]
MerrileesMJ, SodekJ (1992).
Synthesis of TGF-beta 1 by vascular endothelial cells is correlated with cell spreading
Circulating transforming growth factor beta-1: a partial molecular explanation for associations between hypertension, diabetes, obesity, smoking and human disease involving fibrosis
Wyss-CorayT, FengL, MasliahE, RuppeMD, LeeHS, ToggasSM, et al. (1995).
Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-beta 1
Advanced glycation end-products disrupt the blood-brain barrier by stimulating the release of transforming growth factor-beta by pericytes and vascular endothelial growth factor and matrix metalloproteinase-2 by endothelial cells in vitro
Garrido-MartinEM, NguyenHL, CunninghamTA, ChoeSW, JiangZ, ArthurHM, et al. (2014).
Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models--brief report
. Arterioscler Thromb Vasc Biol, 34:2232-2236.
[99]
MahmoudM, AllinsonKR, ZhaiZ, OakenfullR, GhandiP, AdamsRH, et al. (2010).
Pathogenesis of arteriovenous malformations in the absence of endoglin
. Circ Res, 106:1425-1433.
[100]
MahmoudM, UptonPD, ArthurHM (2011).
Angiogenesis regulation by TGFbeta signalling: clues from an inherited vascular disease
. Biochem Soc Trans, 39:1659-1666.
[101]
Tual-ChalotS, MahmoudM, AllinsonKR, RedgraveRE, ZhaiZ, OhSP, et al. (2014).
Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression
The opposing effects of basic fibroblast growth factor and transforming growth factor beta on the regulation of plasminogen activator activity in capillary endothelial cells
PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate
KandasamyM, Couillard-DespresS, RaberKA, StephanM, LehnerB, WinnerB, et al. (2010).
Stem cell quiescence in the hippocampal neurogenic niche is associated with elevated transforming growth factor-beta signaling in an animal model of Huntington disease
KuriyamaN, MizunoT, KitaM, YamadaK, OzakiE, MatsumotoS, et al. (2014).
TGF-beta1 is associated with the progression of intracranial deep white matter lesions: a pilot study with 5 years of magnetic resonance imaging follow-up
Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer's disease
The role of TGF-beta superfamily signaling in neurological disorders
. Acta Biochim Biophys Sin (Shanghai), 50:106-120.
[215]
VogelT, AhrensS, ButtnerN, KrieglsteinK (2010).
Transforming growth factor beta promotes neuronal cell fate of mouse cortical and hippocampal progenitors in vitro and in vivo: identification of Nedd9 as an essential signaling component
Cerebrospinal Fluid Inflammatory Cytokine Aberrations in Alzheimer's Disease, Parkinson's Disease and Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis
LewczukP, RiedererP, O'BryantSE, VerbeekMM, DuboisB, VisserPJ, et al. (2018).
Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: An update of the Consensus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry
KandasamyM, YesudhasA, Poornimai AbiramiGP, RadhakrishnanRK, RoshanSA, JohnsonE, et al. (2019).
Genetic reprogramming of somatic cells into neuroblasts through a co-induction of the doublecortin gene along the Yamanaka factors: A promising approach to model neuroregenerative disorders
Ligand-dependent and -independent interactions with the transforming growth factor type II and I receptor subunits reside in the aminoterminal portion of the ectodomain of the type III subunit
... Dementia describes a set of clinical symptoms affecting cognitive, motor and behavioral aspects, particularly in the geriatric population [1, 2]. Approximately 50 million people suffer from dementia, and its incidence doubles every two decades creating a huge socioeconomic and health care burden worldwide. Vascular Dementia (VaD) contributes to 20-40% of all dementia cases making it the second most common form of dementia after Alzheimer’s disease (AD) [3]. The occurrence of VaD varies among populations. For example, unlike AD, the prevalence of VaD is higher in the Asian compared to the Caucasian population [4]. However, VaD frequently coexists with AD and it is often difficult to distinguish the symptoms between VaD and AD. While AD poses a strong gender bias towards women, the prevalence of VaD is higher in men most likely due to cerebrovascular risk factor being more prevalent in males [5]. Thus far, there are no curative, reparative or regenerative treatments available and therefore, development of therapy for VaD has become an essential but unmet need. ...
... In the early phase, VaD can be hardly differentiated from other cognitive deficits such as mild cognitive impairment [6, 17]. As VaD advances, depending on the brain areas that are affected, clinical manifestations of severe cognitive deficits, loss of memory and motor disorders such as gait instability, psychiatric symptoms such as aggressiveness and depression, loss of bladder function, and increasing difficulties with daily routines become clearly evident [1, 2, 6, 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
Vascular dementia
5
2015
... Dementia describes a set of clinical symptoms affecting cognitive, motor and behavioral aspects, particularly in the geriatric population [1, 2]. Approximately 50 million people suffer from dementia, and its incidence doubles every two decades creating a huge socioeconomic and health care burden worldwide. Vascular Dementia (VaD) contributes to 20-40% of all dementia cases making it the second most common form of dementia after Alzheimer’s disease (AD) [3]. The occurrence of VaD varies among populations. For example, unlike AD, the prevalence of VaD is higher in the Asian compared to the Caucasian population [4]. However, VaD frequently coexists with AD and it is often difficult to distinguish the symptoms between VaD and AD. While AD poses a strong gender bias towards women, the prevalence of VaD is higher in men most likely due to cerebrovascular risk factor being more prevalent in males [5]. Thus far, there are no curative, reparative or regenerative treatments available and therefore, development of therapy for VaD has become an essential but unmet need. ...
... Risk factors for VaD are multifactorial and include aging, illiteracy, genetic predisposition, abnormal conditions and diseases such as hypertension, stroke, diabetes, obesity, coronary infarction, atrial fibrillation, and atypical biochemical blood parameters such as high cholesterol and homocysteine levels. Various lifestyle factors such as smoking, unhealthy diet and physical inactivity have been associated with an increased risk of VaD [2, 6] (Fig. 1). In consequence, reducing or avoiding such risk factors, if possible, is strongly recommended for the prevention of VaD. These include maintenance of healthy blood pressure, prevention or control of diabetes, smoking cessation, maintaining physical fitness and controlling blood cholesterol levels. The rapid increase in cerebrovascular diseases including stroke worldwide suggests that the currently used primary preventive actions against stroke and cardiovascular diseases have very limited efficacy, most likely due to the fact that they function on a voluntary basis with limited self-motivation [7]. This leaves a huge challenge to the scientific community to develop effective therapies to successfully treat VaD. ...
... Based on the unstructured diagnostics criteria, VaD has been classified into different types. In 1997, Konno S et al, proposed eight possible forms of VaD, 1) multi-infarct dementia, 2) post-stroke strategic infarcts dementia, 3) multiple subcortical lacunar lesions, 4) Binswanger's disease or arteriosclerotic subcortical leukoence-phalopathy, 5) mixed type of VaD co-occurring with types 1, 2 and 3, 6) haemorrhagic lesion-induced dementia, 7) subcortical vascular dementia, and 8) mixed type of VaD with AD [8, 9]. The aforementioned sub-types of dementias based on vascular dysfunction include several neuropathological syndromes which are all characterized by a reduced cerebral blood flow (CBF) leading to cognitive dysfunctions and memory loss [2]. Some dementias occur after major and/or recurrent strokes in strategically important brain areas and are often considered a major stroke sequel based on large vessel pathology rather than an independent disease [10, 11]. Less common forms of VaD are various types of vasculitis and inherited diseases that affect vessel integrity, such as CADASIL, or the three-prime repair exonuclease (TREX)-1-related thrombotic microvascular disease [12-15]. The predominant form of VaD is based on cerebral small vessel disease, leading to vascular pathologies in deep penetrating arteries and resulting in numerous minor ischemic-hypoxic lesions (multiple lacunes) in deep cortical areas or subcortical areas [16]. Irrespective of the specific subtype, risks include atherosclerosis of cerebral arteries and cerebral amyloid angiopathy [2, 6] (Fig. 1). ...
... ]. Irrespective of the specific subtype, risks include atherosclerosis of cerebral arteries and cerebral amyloid angiopathy [2, 6] (Fig. 1). ...
... In the early phase, VaD can be hardly differentiated from other cognitive deficits such as mild cognitive impairment [6, 17]. As VaD advances, depending on the brain areas that are affected, clinical manifestations of severe cognitive deficits, loss of memory and motor disorders such as gait instability, psychiatric symptoms such as aggressiveness and depression, loss of bladder function, and increasing difficulties with daily routines become clearly evident [1, 2, 6, 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
Global, regional, and national burden of Alzheimer's disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016
1
2019
... Dementia describes a set of clinical symptoms affecting cognitive, motor and behavioral aspects, particularly in the geriatric population [1, 2]. Approximately 50 million people suffer from dementia, and its incidence doubles every two decades creating a huge socioeconomic and health care burden worldwide. Vascular Dementia (VaD) contributes to 20-40% of all dementia cases making it the second most common form of dementia after Alzheimer’s disease (AD) [3]. The occurrence of VaD varies among populations. For example, unlike AD, the prevalence of VaD is higher in the Asian compared to the Caucasian population [4]. However, VaD frequently coexists with AD and it is often difficult to distinguish the symptoms between VaD and AD. While AD poses a strong gender bias towards women, the prevalence of VaD is higher in men most likely due to cerebrovascular risk factor being more prevalent in males [5]. Thus far, there are no curative, reparative or regenerative treatments available and therefore, development of therapy for VaD has become an essential but unmet need. ...
Global epidemiology of dementia: Alzheimer's and vascular types
1
2014
... Dementia describes a set of clinical symptoms affecting cognitive, motor and behavioral aspects, particularly in the geriatric population [1, 2]. Approximately 50 million people suffer from dementia, and its incidence doubles every two decades creating a huge socioeconomic and health care burden worldwide. Vascular Dementia (VaD) contributes to 20-40% of all dementia cases making it the second most common form of dementia after Alzheimer’s disease (AD) [3]. The occurrence of VaD varies among populations. For example, unlike AD, the prevalence of VaD is higher in the Asian compared to the Caucasian population [4]. However, VaD frequently coexists with AD and it is often difficult to distinguish the symptoms between VaD and AD. While AD poses a strong gender bias towards women, the prevalence of VaD is higher in men most likely due to cerebrovascular risk factor being more prevalent in males [5]. Thus far, there are no curative, reparative or regenerative treatments available and therefore, development of therapy for VaD has become an essential but unmet need. ...
Considering sex and gender in Alzheimer disease and other dementias
1
2016
... Dementia describes a set of clinical symptoms affecting cognitive, motor and behavioral aspects, particularly in the geriatric population [1, 2]. Approximately 50 million people suffer from dementia, and its incidence doubles every two decades creating a huge socioeconomic and health care burden worldwide. Vascular Dementia (VaD) contributes to 20-40% of all dementia cases making it the second most common form of dementia after Alzheimer’s disease (AD) [3]. The occurrence of VaD varies among populations. For example, unlike AD, the prevalence of VaD is higher in the Asian compared to the Caucasian population [4]. However, VaD frequently coexists with AD and it is often difficult to distinguish the symptoms between VaD and AD. While AD poses a strong gender bias towards women, the prevalence of VaD is higher in men most likely due to cerebrovascular risk factor being more prevalent in males [5]. Thus far, there are no curative, reparative or regenerative treatments available and therefore, development of therapy for VaD has become an essential but unmet need. ...
Pathology and pathogenesis of vascular cognitive impairment-a critical update
6
2013
... Risk factors for VaD are multifactorial and include aging, illiteracy, genetic predisposition, abnormal conditions and diseases such as hypertension, stroke, diabetes, obesity, coronary infarction, atrial fibrillation, and atypical biochemical blood parameters such as high cholesterol and homocysteine levels. Various lifestyle factors such as smoking, unhealthy diet and physical inactivity have been associated with an increased risk of VaD [2, 6] (Fig. 1). In consequence, reducing or avoiding such risk factors, if possible, is strongly recommended for the prevention of VaD. These include maintenance of healthy blood pressure, prevention or control of diabetes, smoking cessation, maintaining physical fitness and controlling blood cholesterol levels. The rapid increase in cerebrovascular diseases including stroke worldwide suggests that the currently used primary preventive actions against stroke and cardiovascular diseases have very limited efficacy, most likely due to the fact that they function on a voluntary basis with limited self-motivation [7]. This leaves a huge challenge to the scientific community to develop effective therapies to successfully treat VaD. ...
... Based on the unstructured diagnostics criteria, VaD has been classified into different types. In 1997, Konno S et al, proposed eight possible forms of VaD, 1) multi-infarct dementia, 2) post-stroke strategic infarcts dementia, 3) multiple subcortical lacunar lesions, 4) Binswanger's disease or arteriosclerotic subcortical leukoence-phalopathy, 5) mixed type of VaD co-occurring with types 1, 2 and 3, 6) haemorrhagic lesion-induced dementia, 7) subcortical vascular dementia, and 8) mixed type of VaD with AD [8, 9]. The aforementioned sub-types of dementias based on vascular dysfunction include several neuropathological syndromes which are all characterized by a reduced cerebral blood flow (CBF) leading to cognitive dysfunctions and memory loss [2]. Some dementias occur after major and/or recurrent strokes in strategically important brain areas and are often considered a major stroke sequel based on large vessel pathology rather than an independent disease [10, 11]. Less common forms of VaD are various types of vasculitis and inherited diseases that affect vessel integrity, such as CADASIL, or the three-prime repair exonuclease (TREX)-1-related thrombotic microvascular disease [12-15]. The predominant form of VaD is based on cerebral small vessel disease, leading to vascular pathologies in deep penetrating arteries and resulting in numerous minor ischemic-hypoxic lesions (multiple lacunes) in deep cortical areas or subcortical areas [16]. Irrespective of the specific subtype, risks include atherosclerosis of cerebral arteries and cerebral amyloid angiopathy [2, 6] (Fig. 1). ...
... In the early phase, VaD can be hardly differentiated from other cognitive deficits such as mild cognitive impairment [6, 17]. As VaD advances, depending on the brain areas that are affected, clinical manifestations of severe cognitive deficits, loss of memory and motor disorders such as gait instability, psychiatric symptoms such as aggressiveness and depression, loss of bladder function, and increasing difficulties with daily routines become clearly evident [1, 2, 6, 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
... , 6, 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
... Irrespective of its origin, the pathogenesis of VaD is quite multifaceted. Upon a closer view, this can involve atherosclerosis, amyloid angiopathy, microbleeding and micro-stroke, blood-brain-barrier (BBB) dysfunction and leakage, infiltration and invasion of blood-derived factors such as circulating immune cells, extracellular vesicles and molecules, abnormal vascular remodeling, endothelial cell death and string vessel formation, pericyte responses, fibrosis, astrogliosis, microglia activation, oxidative stress, neuroinflammation, demyelination and white matter lesions, deprivation of synapses and neuronal loss (Fig. 1) [6, 17-19]. Spontaneous intracerebral hemorrhage can be one of the early pathological features of VaD [24-26]. In the lesioned or degenerative brain, BBB disruption leads to an influx of blood molecules, invasion of blood cells and microbial pathogens into the brain and is associated with inflammatory and immune responses, which can initiate multiple pathways of neurodegeneration [27]. The pathogenic mechanisms rendered by BBB breakdown leads to neuronal injury, synaptic dysfunction, loss of neuronal connectivity and neuronal loss, which have been described in neurodegenerative disorders including VaD [28-31]. ...
... The complex pathophysiology in VaD affects neuronal networks involved in behavior, intellectual process, execution and memory, ultimately leading to cognitive deficits [6, 9] (Fig. 1). Considering and accepting this complexity imposes a huge challenge in drug and therapy development for VaD. Most likely, the classical view of a drug with “single mode of action” is inappropriate for a disease like VaD. In consequence, any kind of therapeutic strategy for the diseased brain, including VaD, needs to address a plethora of pathological aspects in a multimodal fashion and needs collectively to include neuroprotection, plasticity, as well as regeneration in the brain. Such an approach most likely needs to be centered around the vascular niche and neurovascular unit, where the various affected cell types meet [32, 33]. ...
Primary prevention of cardiovascular disease through population-wide motivational strategies: insights from using smartphones in stroke prevention
1
2016
... Risk factors for VaD are multifactorial and include aging, illiteracy, genetic predisposition, abnormal conditions and diseases such as hypertension, stroke, diabetes, obesity, coronary infarction, atrial fibrillation, and atypical biochemical blood parameters such as high cholesterol and homocysteine levels. Various lifestyle factors such as smoking, unhealthy diet and physical inactivity have been associated with an increased risk of VaD [2, 6] (Fig. 1). In consequence, reducing or avoiding such risk factors, if possible, is strongly recommended for the prevention of VaD. These include maintenance of healthy blood pressure, prevention or control of diabetes, smoking cessation, maintaining physical fitness and controlling blood cholesterol levels. The rapid increase in cerebrovascular diseases including stroke worldwide suggests that the currently used primary preventive actions against stroke and cardiovascular diseases have very limited efficacy, most likely due to the fact that they function on a voluntary basis with limited self-motivation [7]. This leaves a huge challenge to the scientific community to develop effective therapies to successfully treat VaD. ...
Classification, diagnosis and treatment of vascular dementia
2
1997
... Based on the unstructured diagnostics criteria, VaD has been classified into different types. In 1997, Konno S et al, proposed eight possible forms of VaD, 1) multi-infarct dementia, 2) post-stroke strategic infarcts dementia, 3) multiple subcortical lacunar lesions, 4) Binswanger's disease or arteriosclerotic subcortical leukoence-phalopathy, 5) mixed type of VaD co-occurring with types 1, 2 and 3, 6) haemorrhagic lesion-induced dementia, 7) subcortical vascular dementia, and 8) mixed type of VaD with AD [8, 9]. The aforementioned sub-types of dementias based on vascular dysfunction include several neuropathological syndromes which are all characterized by a reduced cerebral blood flow (CBF) leading to cognitive dysfunctions and memory loss [2]. Some dementias occur after major and/or recurrent strokes in strategically important brain areas and are often considered a major stroke sequel based on large vessel pathology rather than an independent disease [10, 11]. Less common forms of VaD are various types of vasculitis and inherited diseases that affect vessel integrity, such as CADASIL, or the three-prime repair exonuclease (TREX)-1-related thrombotic microvascular disease [12-15]. The predominant form of VaD is based on cerebral small vessel disease, leading to vascular pathologies in deep penetrating arteries and resulting in numerous minor ischemic-hypoxic lesions (multiple lacunes) in deep cortical areas or subcortical areas [16]. Irrespective of the specific subtype, risks include atherosclerosis of cerebral arteries and cerebral amyloid angiopathy [2, 6] (Fig. 1). ...
... In the early phase, VaD can be hardly differentiated from other cognitive deficits such as mild cognitive impairment [6, 17]. As VaD advances, depending on the brain areas that are affected, clinical manifestations of severe cognitive deficits, loss of memory and motor disorders such as gait instability, psychiatric symptoms such as aggressiveness and depression, loss of bladder function, and increasing difficulties with daily routines become clearly evident [1, 2, 6, 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
Models and mechanisms of vascular dementia
2
2015
... Based on the unstructured diagnostics criteria, VaD has been classified into different types. In 1997, Konno S et al, proposed eight possible forms of VaD, 1) multi-infarct dementia, 2) post-stroke strategic infarcts dementia, 3) multiple subcortical lacunar lesions, 4) Binswanger's disease or arteriosclerotic subcortical leukoence-phalopathy, 5) mixed type of VaD co-occurring with types 1, 2 and 3, 6) haemorrhagic lesion-induced dementia, 7) subcortical vascular dementia, and 8) mixed type of VaD with AD [8, 9]. The aforementioned sub-types of dementias based on vascular dysfunction include several neuropathological syndromes which are all characterized by a reduced cerebral blood flow (CBF) leading to cognitive dysfunctions and memory loss [2]. Some dementias occur after major and/or recurrent strokes in strategically important brain areas and are often considered a major stroke sequel based on large vessel pathology rather than an independent disease [10, 11]. Less common forms of VaD are various types of vasculitis and inherited diseases that affect vessel integrity, such as CADASIL, or the three-prime repair exonuclease (TREX)-1-related thrombotic microvascular disease [12-15]. The predominant form of VaD is based on cerebral small vessel disease, leading to vascular pathologies in deep penetrating arteries and resulting in numerous minor ischemic-hypoxic lesions (multiple lacunes) in deep cortical areas or subcortical areas [16]. Irrespective of the specific subtype, risks include atherosclerosis of cerebral arteries and cerebral amyloid angiopathy [2, 6] (Fig. 1). ...
... The complex pathophysiology in VaD affects neuronal networks involved in behavior, intellectual process, execution and memory, ultimately leading to cognitive deficits [6, 9] (Fig. 1). Considering and accepting this complexity imposes a huge challenge in drug and therapy development for VaD. Most likely, the classical view of a drug with “single mode of action” is inappropriate for a disease like VaD. In consequence, any kind of therapeutic strategy for the diseased brain, including VaD, needs to address a plethora of pathological aspects in a multimodal fashion and needs collectively to include neuroprotection, plasticity, as well as regeneration in the brain. Such an approach most likely needs to be centered around the vascular niche and neurovascular unit, where the various affected cell types meet [32, 33]. ...
Increased risk for dementia both before and after stroke: A population-based study in women followed over 44 years
1
2018
... Based on the unstructured diagnostics criteria, VaD has been classified into different types. In 1997, Konno S et al, proposed eight possible forms of VaD, 1) multi-infarct dementia, 2) post-stroke strategic infarcts dementia, 3) multiple subcortical lacunar lesions, 4) Binswanger's disease or arteriosclerotic subcortical leukoence-phalopathy, 5) mixed type of VaD co-occurring with types 1, 2 and 3, 6) haemorrhagic lesion-induced dementia, 7) subcortical vascular dementia, and 8) mixed type of VaD with AD [8, 9]. The aforementioned sub-types of dementias based on vascular dysfunction include several neuropathological syndromes which are all characterized by a reduced cerebral blood flow (CBF) leading to cognitive dysfunctions and memory loss [2]. Some dementias occur after major and/or recurrent strokes in strategically important brain areas and are often considered a major stroke sequel based on large vessel pathology rather than an independent disease [10, 11]. Less common forms of VaD are various types of vasculitis and inherited diseases that affect vessel integrity, such as CADASIL, or the three-prime repair exonuclease (TREX)-1-related thrombotic microvascular disease [12-15]. The predominant form of VaD is based on cerebral small vessel disease, leading to vascular pathologies in deep penetrating arteries and resulting in numerous minor ischemic-hypoxic lesions (multiple lacunes) in deep cortical areas or subcortical areas [16]. Irrespective of the specific subtype, risks include atherosclerosis of cerebral arteries and cerebral amyloid angiopathy [2, 6] (Fig. 1). ...
Incidence and prevalence of dementia associated with transient ischaemic attack and stroke: analysis of the population-based Oxford Vascular Study
1
2019
... Based on the unstructured diagnostics criteria, VaD has been classified into different types. In 1997, Konno S et al, proposed eight possible forms of VaD, 1) multi-infarct dementia, 2) post-stroke strategic infarcts dementia, 3) multiple subcortical lacunar lesions, 4) Binswanger's disease or arteriosclerotic subcortical leukoence-phalopathy, 5) mixed type of VaD co-occurring with types 1, 2 and 3, 6) haemorrhagic lesion-induced dementia, 7) subcortical vascular dementia, and 8) mixed type of VaD with AD [8, 9]. The aforementioned sub-types of dementias based on vascular dysfunction include several neuropathological syndromes which are all characterized by a reduced cerebral blood flow (CBF) leading to cognitive dysfunctions and memory loss [2]. Some dementias occur after major and/or recurrent strokes in strategically important brain areas and are often considered a major stroke sequel based on large vessel pathology rather than an independent disease [10, 11]. Less common forms of VaD are various types of vasculitis and inherited diseases that affect vessel integrity, such as CADASIL, or the three-prime repair exonuclease (TREX)-1-related thrombotic microvascular disease [12-15]. The predominant form of VaD is based on cerebral small vessel disease, leading to vascular pathologies in deep penetrating arteries and resulting in numerous minor ischemic-hypoxic lesions (multiple lacunes) in deep cortical areas or subcortical areas [16]. Irrespective of the specific subtype, risks include atherosclerosis of cerebral arteries and cerebral amyloid angiopathy [2, 6] (Fig. 1). ...
Clinical spectrum of CADASIL: a study of 7 families. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
1
1995
... Based on the unstructured diagnostics criteria, VaD has been classified into different types. In 1997, Konno S et al, proposed eight possible forms of VaD, 1) multi-infarct dementia, 2) post-stroke strategic infarcts dementia, 3) multiple subcortical lacunar lesions, 4) Binswanger's disease or arteriosclerotic subcortical leukoence-phalopathy, 5) mixed type of VaD co-occurring with types 1, 2 and 3, 6) haemorrhagic lesion-induced dementia, 7) subcortical vascular dementia, and 8) mixed type of VaD with AD [8, 9]. The aforementioned sub-types of dementias based on vascular dysfunction include several neuropathological syndromes which are all characterized by a reduced cerebral blood flow (CBF) leading to cognitive dysfunctions and memory loss [2]. Some dementias occur after major and/or recurrent strokes in strategically important brain areas and are often considered a major stroke sequel based on large vessel pathology rather than an independent disease [10, 11]. Less common forms of VaD are various types of vasculitis and inherited diseases that affect vessel integrity, such as CADASIL, or the three-prime repair exonuclease (TREX)-1-related thrombotic microvascular disease [12-15]. The predominant form of VaD is based on cerebral small vessel disease, leading to vascular pathologies in deep penetrating arteries and resulting in numerous minor ischemic-hypoxic lesions (multiple lacunes) in deep cortical areas or subcortical areas [16]. Irrespective of the specific subtype, risks include atherosclerosis of cerebral arteries and cerebral amyloid angiopathy [2, 6] (Fig. 1). ...
TREX1 C-terminal frameshift mutations in the systemic variant of retinal vasculopathy with cerebral leukodystrophy
0
2015
Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia
0
1996
C-terminal truncations in human 3'-5' DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy
1
2007
... Based on the unstructured diagnostics criteria, VaD has been classified into different types. In 1997, Konno S et al, proposed eight possible forms of VaD, 1) multi-infarct dementia, 2) post-stroke strategic infarcts dementia, 3) multiple subcortical lacunar lesions, 4) Binswanger's disease or arteriosclerotic subcortical leukoence-phalopathy, 5) mixed type of VaD co-occurring with types 1, 2 and 3, 6) haemorrhagic lesion-induced dementia, 7) subcortical vascular dementia, and 8) mixed type of VaD with AD [8, 9]. The aforementioned sub-types of dementias based on vascular dysfunction include several neuropathological syndromes which are all characterized by a reduced cerebral blood flow (CBF) leading to cognitive dysfunctions and memory loss [2]. Some dementias occur after major and/or recurrent strokes in strategically important brain areas and are often considered a major stroke sequel based on large vessel pathology rather than an independent disease [10, 11]. Less common forms of VaD are various types of vasculitis and inherited diseases that affect vessel integrity, such as CADASIL, or the three-prime repair exonuclease (TREX)-1-related thrombotic microvascular disease [12-15]. The predominant form of VaD is based on cerebral small vessel disease, leading to vascular pathologies in deep penetrating arteries and resulting in numerous minor ischemic-hypoxic lesions (multiple lacunes) in deep cortical areas or subcortical areas [16]. Irrespective of the specific subtype, risks include atherosclerosis of cerebral arteries and cerebral amyloid angiopathy [2, 6] (Fig. 1). ...
Small vessel disease and subcortical vascular dementia
1
2006
... Based on the unstructured diagnostics criteria, VaD has been classified into different types. In 1997, Konno S et al, proposed eight possible forms of VaD, 1) multi-infarct dementia, 2) post-stroke strategic infarcts dementia, 3) multiple subcortical lacunar lesions, 4) Binswanger's disease or arteriosclerotic subcortical leukoence-phalopathy, 5) mixed type of VaD co-occurring with types 1, 2 and 3, 6) haemorrhagic lesion-induced dementia, 7) subcortical vascular dementia, and 8) mixed type of VaD with AD [8, 9]. The aforementioned sub-types of dementias based on vascular dysfunction include several neuropathological syndromes which are all characterized by a reduced cerebral blood flow (CBF) leading to cognitive dysfunctions and memory loss [2]. Some dementias occur after major and/or recurrent strokes in strategically important brain areas and are often considered a major stroke sequel based on large vessel pathology rather than an independent disease [10, 11]. Less common forms of VaD are various types of vasculitis and inherited diseases that affect vessel integrity, such as CADASIL, or the three-prime repair exonuclease (TREX)-1-related thrombotic microvascular disease [12-15]. The predominant form of VaD is based on cerebral small vessel disease, leading to vascular pathologies in deep penetrating arteries and resulting in numerous minor ischemic-hypoxic lesions (multiple lacunes) in deep cortical areas or subcortical areas [16]. Irrespective of the specific subtype, risks include atherosclerosis of cerebral arteries and cerebral amyloid angiopathy [2, 6] (Fig. 1). ...
The pathology and pathophysiology of vascular dementia
5
2018
... In the early phase, VaD can be hardly differentiated from other cognitive deficits such as mild cognitive impairment [6, 17]. As VaD advances, depending on the brain areas that are affected, clinical manifestations of severe cognitive deficits, loss of memory and motor disorders such as gait instability, psychiatric symptoms such as aggressiveness and depression, loss of bladder function, and increasing difficulties with daily routines become clearly evident [1, 2, 6, 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
... , 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
... ]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
... Irrespective of its origin, the pathogenesis of VaD is quite multifaceted. Upon a closer view, this can involve atherosclerosis, amyloid angiopathy, microbleeding and micro-stroke, blood-brain-barrier (BBB) dysfunction and leakage, infiltration and invasion of blood-derived factors such as circulating immune cells, extracellular vesicles and molecules, abnormal vascular remodeling, endothelial cell death and string vessel formation, pericyte responses, fibrosis, astrogliosis, microglia activation, oxidative stress, neuroinflammation, demyelination and white matter lesions, deprivation of synapses and neuronal loss (Fig. 1) [6, 17-19]. Spontaneous intracerebral hemorrhage can be one of the early pathological features of VaD [24-26]. In the lesioned or degenerative brain, BBB disruption leads to an influx of blood molecules, invasion of blood cells and microbial pathogens into the brain and is associated with inflammatory and immune responses, which can initiate multiple pathways of neurodegeneration [27]. The pathogenic mechanisms rendered by BBB breakdown leads to neuronal injury, synaptic dysfunction, loss of neuronal connectivity and neuronal loss, which have been described in neurodegenerative disorders including VaD [28-31]. ...
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Pathophysiology of vascular dementia
1
2009
... In the early phase, VaD can be hardly differentiated from other cognitive deficits such as mild cognitive impairment [6, 17]. As VaD advances, depending on the brain areas that are affected, clinical manifestations of severe cognitive deficits, loss of memory and motor disorders such as gait instability, psychiatric symptoms such as aggressiveness and depression, loss of bladder function, and increasing difficulties with daily routines become clearly evident [1, 2, 6, 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
Vascular cognitive disorder. A biological and clinical overview
2
2010
... In the early phase, VaD can be hardly differentiated from other cognitive deficits such as mild cognitive impairment [6, 17]. As VaD advances, depending on the brain areas that are affected, clinical manifestations of severe cognitive deficits, loss of memory and motor disorders such as gait instability, psychiatric symptoms such as aggressiveness and depression, loss of bladder function, and increasing difficulties with daily routines become clearly evident [1, 2, 6, 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
... Irrespective of its origin, the pathogenesis of VaD is quite multifaceted. Upon a closer view, this can involve atherosclerosis, amyloid angiopathy, microbleeding and micro-stroke, blood-brain-barrier (BBB) dysfunction and leakage, infiltration and invasion of blood-derived factors such as circulating immune cells, extracellular vesicles and molecules, abnormal vascular remodeling, endothelial cell death and string vessel formation, pericyte responses, fibrosis, astrogliosis, microglia activation, oxidative stress, neuroinflammation, demyelination and white matter lesions, deprivation of synapses and neuronal loss (Fig. 1) [6, 17-19]. Spontaneous intracerebral hemorrhage can be one of the early pathological features of VaD [24-26]. In the lesioned or degenerative brain, BBB disruption leads to an influx of blood molecules, invasion of blood cells and microbial pathogens into the brain and is associated with inflammatory and immune responses, which can initiate multiple pathways of neurodegeneration [27]. The pathogenic mechanisms rendered by BBB breakdown leads to neuronal injury, synaptic dysfunction, loss of neuronal connectivity and neuronal loss, which have been described in neurodegenerative disorders including VaD [28-31]. ...
Neuroimaging of vascular dementia
1
2014
... In the early phase, VaD can be hardly differentiated from other cognitive deficits such as mild cognitive impairment [6, 17]. As VaD advances, depending on the brain areas that are affected, clinical manifestations of severe cognitive deficits, loss of memory and motor disorders such as gait instability, psychiatric symptoms such as aggressiveness and depression, loss of bladder function, and increasing difficulties with daily routines become clearly evident [1, 2, 6, 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
Identifying preclinical vascular dementia in symptomatic small vessel disease using MRI
0
2018
Ultrasound and dynamic functional imaging in vascular cognitive impairment and Alzheimer's disease
0
2017
MRI and CT in the diagnosis of vascular dementia
1
2004
... In the early phase, VaD can be hardly differentiated from other cognitive deficits such as mild cognitive impairment [6, 17]. As VaD advances, depending on the brain areas that are affected, clinical manifestations of severe cognitive deficits, loss of memory and motor disorders such as gait instability, psychiatric symptoms such as aggressiveness and depression, loss of bladder function, and increasing difficulties with daily routines become clearly evident [1, 2, 6, 17, 18]. Overlapping symptoms of VaD with other dementias and its occurrence with mixed pathologies creates a huge challenge for accurate diagnosis [17, 19]. Besides the obvious clinical symptoms, diagnosis of VaD requires the proof of impaired cerebral blood flow in the brain and vascular imaging such as MRI, CT, angiography and carotid ultrasound [8, 20-23]. ...
Dementia risk after spontaneous intracerebral haemorrhage: a prospective cohort study
1
2016
... Irrespective of its origin, the pathogenesis of VaD is quite multifaceted. Upon a closer view, this can involve atherosclerosis, amyloid angiopathy, microbleeding and micro-stroke, blood-brain-barrier (BBB) dysfunction and leakage, infiltration and invasion of blood-derived factors such as circulating immune cells, extracellular vesicles and molecules, abnormal vascular remodeling, endothelial cell death and string vessel formation, pericyte responses, fibrosis, astrogliosis, microglia activation, oxidative stress, neuroinflammation, demyelination and white matter lesions, deprivation of synapses and neuronal loss (Fig. 1) [6, 17-19]. Spontaneous intracerebral hemorrhage can be one of the early pathological features of VaD [24-26]. In the lesioned or degenerative brain, BBB disruption leads to an influx of blood molecules, invasion of blood cells and microbial pathogens into the brain and is associated with inflammatory and immune responses, which can initiate multiple pathways of neurodegeneration [27]. The pathogenic mechanisms rendered by BBB breakdown leads to neuronal injury, synaptic dysfunction, loss of neuronal connectivity and neuronal loss, which have been described in neurodegenerative disorders including VaD [28-31]. ...
Toll-Like Receptor 4/MyD88-Mediated Signaling of Hepcidin Expression Causing Brain Iron Accumulation, Oxidative Injury, and Cognitive Impairment After Intracerebral Hemorrhage
0
2016
Early Cognitive Impairment after Intracerebral Hemorrhage in the INTERACT1 Study
1
2017
... Irrespective of its origin, the pathogenesis of VaD is quite multifaceted. Upon a closer view, this can involve atherosclerosis, amyloid angiopathy, microbleeding and micro-stroke, blood-brain-barrier (BBB) dysfunction and leakage, infiltration and invasion of blood-derived factors such as circulating immune cells, extracellular vesicles and molecules, abnormal vascular remodeling, endothelial cell death and string vessel formation, pericyte responses, fibrosis, astrogliosis, microglia activation, oxidative stress, neuroinflammation, demyelination and white matter lesions, deprivation of synapses and neuronal loss (Fig. 1) [6, 17-19]. Spontaneous intracerebral hemorrhage can be one of the early pathological features of VaD [24-26]. In the lesioned or degenerative brain, BBB disruption leads to an influx of blood molecules, invasion of blood cells and microbial pathogens into the brain and is associated with inflammatory and immune responses, which can initiate multiple pathways of neurodegeneration [27]. The pathogenic mechanisms rendered by BBB breakdown leads to neuronal injury, synaptic dysfunction, loss of neuronal connectivity and neuronal loss, which have been described in neurodegenerative disorders including VaD [28-31]. ...
Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders
1
2018
... Irrespective of its origin, the pathogenesis of VaD is quite multifaceted. Upon a closer view, this can involve atherosclerosis, amyloid angiopathy, microbleeding and micro-stroke, blood-brain-barrier (BBB) dysfunction and leakage, infiltration and invasion of blood-derived factors such as circulating immune cells, extracellular vesicles and molecules, abnormal vascular remodeling, endothelial cell death and string vessel formation, pericyte responses, fibrosis, astrogliosis, microglia activation, oxidative stress, neuroinflammation, demyelination and white matter lesions, deprivation of synapses and neuronal loss (Fig. 1) [6, 17-19]. Spontaneous intracerebral hemorrhage can be one of the early pathological features of VaD [24-26]. In the lesioned or degenerative brain, BBB disruption leads to an influx of blood molecules, invasion of blood cells and microbial pathogens into the brain and is associated with inflammatory and immune responses, which can initiate multiple pathways of neurodegeneration [27]. The pathogenic mechanisms rendered by BBB breakdown leads to neuronal injury, synaptic dysfunction, loss of neuronal connectivity and neuronal loss, which have been described in neurodegenerative disorders including VaD [28-31]. ...
Blood-brain barrier damage in vascular dementia
1
2016
... Irrespective of its origin, the pathogenesis of VaD is quite multifaceted. Upon a closer view, this can involve atherosclerosis, amyloid angiopathy, microbleeding and micro-stroke, blood-brain-barrier (BBB) dysfunction and leakage, infiltration and invasion of blood-derived factors such as circulating immune cells, extracellular vesicles and molecules, abnormal vascular remodeling, endothelial cell death and string vessel formation, pericyte responses, fibrosis, astrogliosis, microglia activation, oxidative stress, neuroinflammation, demyelination and white matter lesions, deprivation of synapses and neuronal loss (Fig. 1) [6, 17-19]. Spontaneous intracerebral hemorrhage can be one of the early pathological features of VaD [24-26]. In the lesioned or degenerative brain, BBB disruption leads to an influx of blood molecules, invasion of blood cells and microbial pathogens into the brain and is associated with inflammatory and immune responses, which can initiate multiple pathways of neurodegeneration [27]. The pathogenic mechanisms rendered by BBB breakdown leads to neuronal injury, synaptic dysfunction, loss of neuronal connectivity and neuronal loss, which have been described in neurodegenerative disorders including VaD [28-31]. ...
Blood brain barrier function in vascular dementia
0
1990
Biochemical markers in vascular cognitive impairment associated with subcortical small vessel disease - A consensus report
1
2017
... On the molecular and genetic level, a few candidates for pathogenesis in VaD have been discussed. For example, polymorphisms and differential expression analyses have identified apolipoprotein E (APOE), methylene-tetra hydrofolate reductase (MTHFR), paraoxonase 1 (PON1), tumor necrosis factor (TNF)-alpha, vascular endothelial growth factor (VEGF), and transforming growth factor (TGF)-β1 as potential candidate factors involved in the pathogenesis of VaD [30, 34]. Among them, TGF-β1 has gained a remarkable scientific interest as it plays a crucial role in angiogenesis, neuroprotection, neuroimmune functions, neural regeneration and synaptic plasticity, which are responsible for cognitive functions in the physiological state [35-38]. ...
Establishment and Dysfunction of the Blood-Brain Barrier
1
2015
... Irrespective of its origin, the pathogenesis of VaD is quite multifaceted. Upon a closer view, this can involve atherosclerosis, amyloid angiopathy, microbleeding and micro-stroke, blood-brain-barrier (BBB) dysfunction and leakage, infiltration and invasion of blood-derived factors such as circulating immune cells, extracellular vesicles and molecules, abnormal vascular remodeling, endothelial cell death and string vessel formation, pericyte responses, fibrosis, astrogliosis, microglia activation, oxidative stress, neuroinflammation, demyelination and white matter lesions, deprivation of synapses and neuronal loss (Fig. 1) [6, 17-19]. Spontaneous intracerebral hemorrhage can be one of the early pathological features of VaD [24-26]. In the lesioned or degenerative brain, BBB disruption leads to an influx of blood molecules, invasion of blood cells and microbial pathogens into the brain and is associated with inflammatory and immune responses, which can initiate multiple pathways of neurodegeneration [27]. The pathogenic mechanisms rendered by BBB breakdown leads to neuronal injury, synaptic dysfunction, loss of neuronal connectivity and neuronal loss, which have been described in neurodegenerative disorders including VaD [28-31]. ...
Recent progress in translational research on neurovascular and neurodegenerative disorders
1
2017
... The complex pathophysiology in VaD affects neuronal networks involved in behavior, intellectual process, execution and memory, ultimately leading to cognitive deficits [6, 9] (Fig. 1). Considering and accepting this complexity imposes a huge challenge in drug and therapy development for VaD. Most likely, the classical view of a drug with “single mode of action” is inappropriate for a disease like VaD. In consequence, any kind of therapeutic strategy for the diseased brain, including VaD, needs to address a plethora of pathological aspects in a multimodal fashion and needs collectively to include neuroprotection, plasticity, as well as regeneration in the brain. Such an approach most likely needs to be centered around the vascular niche and neurovascular unit, where the various affected cell types meet [32, 33]. ...
Editorial: The Vascular Niche in Tissue Repair: A Therapeutic Target for Regeneration
3
2017
... The complex pathophysiology in VaD affects neuronal networks involved in behavior, intellectual process, execution and memory, ultimately leading to cognitive deficits [6, 9] (Fig. 1). Considering and accepting this complexity imposes a huge challenge in drug and therapy development for VaD. Most likely, the classical view of a drug with “single mode of action” is inappropriate for a disease like VaD. In consequence, any kind of therapeutic strategy for the diseased brain, including VaD, needs to address a plethora of pathological aspects in a multimodal fashion and needs collectively to include neuroprotection, plasticity, as well as regeneration in the brain. Such an approach most likely needs to be centered around the vascular niche and neurovascular unit, where the various affected cell types meet [32, 33]. ...
... Pericytes are multifunctional cells which ensheath the blood vessel and contribute to the regulation of capillary diameter, vasoconstriction and vasodilation, capillary blood flow and extracellular matrix protein secretion [33, 137, 138]. Moreover, pericytes have an immune-modulatory role in the brain [139]. With reference to the origin of the pericytes, both the mesenchymal and neural crest cells have been proposed to give rise to pericytes in many organs. It has recently been reported that pericytes play important roles in the homeostasis of the brain [140-142]. Pericytes are positioned within the BBB between endothelial cells, astrocytes and neurons [138]. Thus, pericytes can integrate and process signals from their adjacent cells to mediate diverse functional responses that are important for brain function in health and disease, including the regulation of the BBB permeability, angiogenesis, clearance of toxic metabolites, phagocytotic activity, capillary hemodynamic responses and neuroinflammation. ...
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
The gene encoding transforming growth factor beta 1 confers risk of ischemic stroke and vascular dementia
3
2006
... On the molecular and genetic level, a few candidates for pathogenesis in VaD have been discussed. For example, polymorphisms and differential expression analyses have identified apolipoprotein E (APOE), methylene-tetra hydrofolate reductase (MTHFR), paraoxonase 1 (PON1), tumor necrosis factor (TNF)-alpha, vascular endothelial growth factor (VEGF), and transforming growth factor (TGF)-β1 as potential candidate factors involved in the pathogenesis of VaD [30, 34]. Among them, TGF-β1 has gained a remarkable scientific interest as it plays a crucial role in angiogenesis, neuroprotection, neuroimmune functions, neural regeneration and synaptic plasticity, which are responsible for cognitive functions in the physiological state [35-38]. ...
... Elevated levels of TGF-β1 in response to cerebro-vascular pathologies, neurodegeneration, neuro-inflammation, and neuroregenerative failure appear to be involved in the progression of dementia [34, 58-62]. Also, genetic variations in TGF-β and TGF-β receptors are considered as the prominent risk factors for VaD. Thus, TGF-β signaling is a therapeutic target candidate in various forms of cognitive disorders. Detailed information on the possible involvement of TGF-β in VaD is limited. In the present review, we focus on TGF-β and its potential role in VaD, in particular, due to its multimodal effects in the brain and neuropathogenesis. ...
... Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73]. ...
TGF-beta in neural stem cells and in tumors of the central nervous system
6
2008
... On the molecular and genetic level, a few candidates for pathogenesis in VaD have been discussed. For example, polymorphisms and differential expression analyses have identified apolipoprotein E (APOE), methylene-tetra hydrofolate reductase (MTHFR), paraoxonase 1 (PON1), tumor necrosis factor (TNF)-alpha, vascular endothelial growth factor (VEGF), and transforming growth factor (TGF)-β1 as potential candidate factors involved in the pathogenesis of VaD [30, 34]. Among them, TGF-β1 has gained a remarkable scientific interest as it plays a crucial role in angiogenesis, neuroprotection, neuroimmune functions, neural regeneration and synaptic plasticity, which are responsible for cognitive functions in the physiological state [35-38]. ...
... TGF-β proteins (TGF-β 1, 2, and 3) are secreted in an inactive form by various cells in various tissues and organs [35]. Thrombospondin (TSP)-1, plasmin, and matrix metalloproteinase (MMP)-2 and -9 trigger the activation process of TGF-β [39]. Besides, the generation of reactive oxygen species (ROS) and the accumulation of an acid milieu in damaged tissues facilitate the activation of TGF-β, thereby leading to the release of TGF-β ligands for the receptor-mediated activation of Smads [40-43]. In particular, TGF-β induces the phosphorylation of Smad2 and Smad3 that bind to Smad4 in the cytoplasm and translocates it into the nucleus [35, 38, 44]. This heteromeric complex acts as a transcription factor to control the expression of various target genes. Thus, TGF-β facilitates transcriptional regulation to create a complex and synergistic higher-order signaling network in various complex biological mechanisms [35, 45] (Fig. 2). ...
... ]. In particular, TGF-β induces the phosphorylation of Smad2 and Smad3 that bind to Smad4 in the cytoplasm and translocates it into the nucleus [35, 38, 44]. This heteromeric complex acts as a transcription factor to control the expression of various target genes. Thus, TGF-β facilitates transcriptional regulation to create a complex and synergistic higher-order signaling network in various complex biological mechanisms [35, 45] (Fig. 2). ...
... ]. This heteromeric complex acts as a transcription factor to control the expression of various target genes. Thus, TGF-β facilitates transcriptional regulation to create a complex and synergistic higher-order signaling network in various complex biological mechanisms [35, 45] (Fig. 2). ...
... TGF-β1 regulates cell proliferation, differentiation, maturation and survival of various neuronal and non-neuronal cell types in the brain [38, 46]. Also, it regulates angiogenesis, BBB integrity, neuroinflammation, neuroplasticity and neuronal function [35, 38, 47-50]. The choroid plexus, as well as astrocytes, microglia, and endothelial cells, have been the major sources of TGF-β1 in the brain [51-53]. In the brain, TGF-β1 is typically elevated after acute brain injury and in chronic neurodegenerative pathologies, where it is the main driver of the astroglial scar formation, fibrosis and sclerosis [54-57]. ...
... In the context of cancer, increased TGF-β facilitates metastasis of tumor cells through the down-regulation of junctional adhesion and tight junction proteins upon the induction of matrix metalloproteases (MMPs) [35, 93]. Similarly, in neuroinflammatory conditions, down-regulation of the expression of junctional adhesion and tight junction proteins in the blood vessel and increased expression of MMPs in association with abnormal TGF-β signaling primes the leakage of BBB, which in turn leads to degeneration of endothelial cells, pericytes drifting and string vessels in the neurovascular systems [94]. Based on the observation from the ablation of the TGF-β signaling pathway in pericytes through retroviral-mediated expression of a truncated C-terminal intracellular kinase domain of TGF-βR2, Sieczkiewicz GJ et al. have described that endothelial cell-specific secretion of TGF-β inhibits the mitotic activities of pericytes through the activation of pericyte contractile protein leading to the BBB breakdown [95]. ...
TGF-beta1 pathway as a new target for neuroprotection in Alzheimer's disease
0
2011
TGF-beta 1 protects hippocampal neurons against degeneration caused by transient global ischemia. Dose-response relationship and potential neuroprotective mechanisms
0
1996
TGF-beta signalling in the adult neurogenic niche promotes stem cell quiescence as well as generation of new neurons
12
2014
... On the molecular and genetic level, a few candidates for pathogenesis in VaD have been discussed. For example, polymorphisms and differential expression analyses have identified apolipoprotein E (APOE), methylene-tetra hydrofolate reductase (MTHFR), paraoxonase 1 (PON1), tumor necrosis factor (TNF)-alpha, vascular endothelial growth factor (VEGF), and transforming growth factor (TGF)-β1 as potential candidate factors involved in the pathogenesis of VaD [30, 34]. Among them, TGF-β1 has gained a remarkable scientific interest as it plays a crucial role in angiogenesis, neuroprotection, neuroimmune functions, neural regeneration and synaptic plasticity, which are responsible for cognitive functions in the physiological state [35-38]. ...
... TGF-β proteins (TGF-β 1, 2, and 3) are secreted in an inactive form by various cells in various tissues and organs [35]. Thrombospondin (TSP)-1, plasmin, and matrix metalloproteinase (MMP)-2 and -9 trigger the activation process of TGF-β [39]. Besides, the generation of reactive oxygen species (ROS) and the accumulation of an acid milieu in damaged tissues facilitate the activation of TGF-β, thereby leading to the release of TGF-β ligands for the receptor-mediated activation of Smads [40-43]. In particular, TGF-β induces the phosphorylation of Smad2 and Smad3 that bind to Smad4 in the cytoplasm and translocates it into the nucleus [35, 38, 44]. This heteromeric complex acts as a transcription factor to control the expression of various target genes. Thus, TGF-β facilitates transcriptional regulation to create a complex and synergistic higher-order signaling network in various complex biological mechanisms [35, 45] (Fig. 2). ...
... TGF-β1 regulates cell proliferation, differentiation, maturation and survival of various neuronal and non-neuronal cell types in the brain [38, 46]. Also, it regulates angiogenesis, BBB integrity, neuroinflammation, neuroplasticity and neuronal function [35, 38, 47-50]. The choroid plexus, as well as astrocytes, microglia, and endothelial cells, have been the major sources of TGF-β1 in the brain [51-53]. In the brain, TGF-β1 is typically elevated after acute brain injury and in chronic neurodegenerative pathologies, where it is the main driver of the astroglial scar formation, fibrosis and sclerosis [54-57]. ...
... , 38, 47-50]. The choroid plexus, as well as astrocytes, microglia, and endothelial cells, have been the major sources of TGF-β1 in the brain [51-53]. In the brain, TGF-β1 is typically elevated after acute brain injury and in chronic neurodegenerative pathologies, where it is the main driver of the astroglial scar formation, fibrosis and sclerosis [54-57]. ...
... Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... ]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... ]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... ]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... ]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... ]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... ]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
LTBP-1 blockade in dioxin receptor-null mouse embryo fibroblasts decreases TGF-beta activity: Role of extracellular proteases plasmin and elastase
1
2006
... TGF-β proteins (TGF-β 1, 2, and 3) are secreted in an inactive form by various cells in various tissues and organs [35]. Thrombospondin (TSP)-1, plasmin, and matrix metalloproteinase (MMP)-2 and -9 trigger the activation process of TGF-β [39]. Besides, the generation of reactive oxygen species (ROS) and the accumulation of an acid milieu in damaged tissues facilitate the activation of TGF-β, thereby leading to the release of TGF-β ligands for the receptor-mediated activation of Smads [40-43]. In particular, TGF-β induces the phosphorylation of Smad2 and Smad3 that bind to Smad4 in the cytoplasm and translocates it into the nucleus [35, 38, 44]. This heteromeric complex acts as a transcription factor to control the expression of various target genes. Thus, TGF-β facilitates transcriptional regulation to create a complex and synergistic higher-order signaling network in various complex biological mechanisms [35, 45] (Fig. 2). ...
Making sense of latent TGFbeta activation
1
2003
... TGF-β proteins (TGF-β 1, 2, and 3) are secreted in an inactive form by various cells in various tissues and organs [35]. Thrombospondin (TSP)-1, plasmin, and matrix metalloproteinase (MMP)-2 and -9 trigger the activation process of TGF-β [39]. Besides, the generation of reactive oxygen species (ROS) and the accumulation of an acid milieu in damaged tissues facilitate the activation of TGF-β, thereby leading to the release of TGF-β ligands for the receptor-mediated activation of Smads [40-43]. In particular, TGF-β induces the phosphorylation of Smad2 and Smad3 that bind to Smad4 in the cytoplasm and translocates it into the nucleus [35, 38, 44]. This heteromeric complex acts as a transcription factor to control the expression of various target genes. Thus, TGF-β facilitates transcriptional regulation to create a complex and synergistic higher-order signaling network in various complex biological mechanisms [35, 45] (Fig. 2). ...
Reciprocal regulation of TGF-beta and reactive oxygen species: A perverse cycle for fibrosis
0
2015
Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium
0
1988
Characterization of latent transforming growth factor-beta from human seminal plasma
1
1995
... TGF-β proteins (TGF-β 1, 2, and 3) are secreted in an inactive form by various cells in various tissues and organs [35]. Thrombospondin (TSP)-1, plasmin, and matrix metalloproteinase (MMP)-2 and -9 trigger the activation process of TGF-β [39]. Besides, the generation of reactive oxygen species (ROS) and the accumulation of an acid milieu in damaged tissues facilitate the activation of TGF-β, thereby leading to the release of TGF-β ligands for the receptor-mediated activation of Smads [40-43]. In particular, TGF-β induces the phosphorylation of Smad2 and Smad3 that bind to Smad4 in the cytoplasm and translocates it into the nucleus [35, 38, 44]. This heteromeric complex acts as a transcription factor to control the expression of various target genes. Thus, TGF-β facilitates transcriptional regulation to create a complex and synergistic higher-order signaling network in various complex biological mechanisms [35, 45] (Fig. 2). ...
TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility
1
2007
... TGF-β proteins (TGF-β 1, 2, and 3) are secreted in an inactive form by various cells in various tissues and organs [35]. Thrombospondin (TSP)-1, plasmin, and matrix metalloproteinase (MMP)-2 and -9 trigger the activation process of TGF-β [39]. Besides, the generation of reactive oxygen species (ROS) and the accumulation of an acid milieu in damaged tissues facilitate the activation of TGF-β, thereby leading to the release of TGF-β ligands for the receptor-mediated activation of Smads [40-43]. In particular, TGF-β induces the phosphorylation of Smad2 and Smad3 that bind to Smad4 in the cytoplasm and translocates it into the nucleus [35, 38, 44]. This heteromeric complex acts as a transcription factor to control the expression of various target genes. Thus, TGF-β facilitates transcriptional regulation to create a complex and synergistic higher-order signaling network in various complex biological mechanisms [35, 45] (Fig. 2). ...
Signaling Cross Talk between TGF-beta/Smad and Other Signaling Pathways
1
2017
... TGF-β proteins (TGF-β 1, 2, and 3) are secreted in an inactive form by various cells in various tissues and organs [35]. Thrombospondin (TSP)-1, plasmin, and matrix metalloproteinase (MMP)-2 and -9 trigger the activation process of TGF-β [39]. Besides, the generation of reactive oxygen species (ROS) and the accumulation of an acid milieu in damaged tissues facilitate the activation of TGF-β, thereby leading to the release of TGF-β ligands for the receptor-mediated activation of Smads [40-43]. In particular, TGF-β induces the phosphorylation of Smad2 and Smad3 that bind to Smad4 in the cytoplasm and translocates it into the nucleus [35, 38, 44]. This heteromeric complex acts as a transcription factor to control the expression of various target genes. Thus, TGF-β facilitates transcriptional regulation to create a complex and synergistic higher-order signaling network in various complex biological mechanisms [35, 45] (Fig. 2). ...
TGF-beta and the regulation of neuron survival and death
1
2002
... TGF-β1 regulates cell proliferation, differentiation, maturation and survival of various neuronal and non-neuronal cell types in the brain [38, 46]. Also, it regulates angiogenesis, BBB integrity, neuroinflammation, neuroplasticity and neuronal function [35, 38, 47-50]. The choroid plexus, as well as astrocytes, microglia, and endothelial cells, have been the major sources of TGF-β1 in the brain [51-53]. In the brain, TGF-β1 is typically elevated after acute brain injury and in chronic neurodegenerative pathologies, where it is the main driver of the astroglial scar formation, fibrosis and sclerosis [54-57]. ...
... TGF-β1 regulates cell proliferation, differentiation, maturation and survival of various neuronal and non-neuronal cell types in the brain [38, 46]. Also, it regulates angiogenesis, BBB integrity, neuroinflammation, neuroplasticity and neuronal function [35, 38, 47-50]. The choroid plexus, as well as astrocytes, microglia, and endothelial cells, have been the major sources of TGF-β1 in the brain [51-53]. In the brain, TGF-β1 is typically elevated after acute brain injury and in chronic neurodegenerative pathologies, where it is the main driver of the astroglial scar formation, fibrosis and sclerosis [54-57]. ...
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Transforming growth factor-beta 1 signaling regulates neuroinflammation and apoptosis in mild traumatic brain injury
0
2017
Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain
0
2009
Anti-inflammatory and pro-inflammatory roles of TGF-beta, IL-10, and IL-22 in immunity and autoimmunity
1
2009
... TGF-β1 regulates cell proliferation, differentiation, maturation and survival of various neuronal and non-neuronal cell types in the brain [38, 46]. Also, it regulates angiogenesis, BBB integrity, neuroinflammation, neuroplasticity and neuronal function [35, 38, 47-50]. The choroid plexus, as well as astrocytes, microglia, and endothelial cells, have been the major sources of TGF-β1 in the brain [51-53]. In the brain, TGF-β1 is typically elevated after acute brain injury and in chronic neurodegenerative pathologies, where it is the main driver of the astroglial scar formation, fibrosis and sclerosis [54-57]. ...
Astrocyte Transforming Growth Factor Beta 1 Protects Synapses against Abeta Oligomers in Alzheimer's Disease Model
2
2017
... TGF-β1 regulates cell proliferation, differentiation, maturation and survival of various neuronal and non-neuronal cell types in the brain [38, 46]. Also, it regulates angiogenesis, BBB integrity, neuroinflammation, neuroplasticity and neuronal function [35, 38, 47-50]. The choroid plexus, as well as astrocytes, microglia, and endothelial cells, have been the major sources of TGF-β1 in the brain [51-53]. In the brain, TGF-β1 is typically elevated after acute brain injury and in chronic neurodegenerative pathologies, where it is the main driver of the astroglial scar formation, fibrosis and sclerosis [54-57]. ...
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
Transforming growth factor-beta1 in the cerebrospinal fluid of patients with subarachnoid hemorrhage: titers derived from exogenous and endogenous sources
0
2001
Synthesis of TGF-beta 1 by vascular endothelial cells is correlated with cell spreading
1
1992
... TGF-β1 regulates cell proliferation, differentiation, maturation and survival of various neuronal and non-neuronal cell types in the brain [38, 46]. Also, it regulates angiogenesis, BBB integrity, neuroinflammation, neuroplasticity and neuronal function [35, 38, 47-50]. The choroid plexus, as well as astrocytes, microglia, and endothelial cells, have been the major sources of TGF-β1 in the brain [51-53]. In the brain, TGF-β1 is typically elevated after acute brain injury and in chronic neurodegenerative pathologies, where it is the main driver of the astroglial scar formation, fibrosis and sclerosis [54-57]. ...
TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke
3
2010
... TGF-β1 regulates cell proliferation, differentiation, maturation and survival of various neuronal and non-neuronal cell types in the brain [38, 46]. Also, it regulates angiogenesis, BBB integrity, neuroinflammation, neuroplasticity and neuronal function [35, 38, 47-50]. The choroid plexus, as well as astrocytes, microglia, and endothelial cells, have been the major sources of TGF-β1 in the brain [51-53]. In the brain, TGF-β1 is typically elevated after acute brain injury and in chronic neurodegenerative pathologies, where it is the main driver of the astroglial scar formation, fibrosis and sclerosis [54-57]. ...
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
... Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
Reciprocal modulation between microglia and astrocyte in reactive gliosis following the CNS injury
0
2013
Transforming growth factor-beta 1 differentially regulates proliferation and MHC class-II antigen expression in forebrain and brainstem astrocyte primary cultures
0
1992
TGF-beta: the master regulator of fibrosis
1
2016
... TGF-β1 regulates cell proliferation, differentiation, maturation and survival of various neuronal and non-neuronal cell types in the brain [38, 46]. Also, it regulates angiogenesis, BBB integrity, neuroinflammation, neuroplasticity and neuronal function [35, 38, 47-50]. The choroid plexus, as well as astrocytes, microglia, and endothelial cells, have been the major sources of TGF-β1 in the brain [51-53]. In the brain, TGF-β1 is typically elevated after acute brain injury and in chronic neurodegenerative pathologies, where it is the main driver of the astroglial scar formation, fibrosis and sclerosis [54-57]. ...
Transforming Growth Factor-Beta Signaling in the Neural Stem Cell Niche: A Therapeutic Target for Huntington's Disease
7
2011
... Elevated levels of TGF-β1 in response to cerebro-vascular pathologies, neurodegeneration, neuro-inflammation, and neuroregenerative failure appear to be involved in the progression of dementia [34, 58-62]. Also, genetic variations in TGF-β and TGF-β receptors are considered as the prominent risk factors for VaD. Thus, TGF-β signaling is a therapeutic target candidate in various forms of cognitive disorders. Detailed information on the possible involvement of TGF-β in VaD is limited. In the present review, we focus on TGF-β and its potential role in VaD, in particular, due to its multimodal effects in the brain and neuropathogenesis. ...
... Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... , 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... ]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... ]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... ]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
New Insight Into the Pathogenesis of Cerebral Small-Vessel Diseases
1
2017
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer's disease and vascular dementia
1
2002
... Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73]. ...
Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology
2
2006
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... Several studies have indicated that the induction of TGF-β during pathogenesis exerts a defensive role in preventing neurodegeneration [219]. Reduced expression of TGF-β and deficient Smad signaling in neurons induces neurodegeneration in the terminal stage of the disease [61]. The reduced neurogenesis seen in the brains of some models of AD could provide a possible explanation for the reduction in TGF-β mediated neurodegeneration in cognitive centers of the pathological brain. Recently, reactive neuroblasts in the diseased brain have been proposed to acquire properties of non-neuronal cells such as microglia, and thus these cells might represent a potential source of TGF-β in pathological brains [223, 253]. The robust activation, proliferation and circulation of microglia, reactive astrocytes and neuroblasts found in early pathogenesis tend to be diminished in the late phases of the diseases [227, 254]. Thus, it can be hypothesized that the overall loss of neuroblasts, non-neuronal and neuronal cells in the brain in the late phase of the disease might lead to the reduced level of TGF-β, thereby fostering neurodegeneration and memory loss. ...
Chronic overproduction of transforming growth factor-beta1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice
3
2000
... Elevated levels of TGF-β1 in response to cerebro-vascular pathologies, neurodegeneration, neuro-inflammation, and neuroregenerative failure appear to be involved in the progression of dementia [34, 58-62]. Also, genetic variations in TGF-β and TGF-β receptors are considered as the prominent risk factors for VaD. Thus, TGF-β signaling is a therapeutic target candidate in various forms of cognitive disorders. Detailed information on the possible involvement of TGF-β in VaD is limited. In the present review, we focus on TGF-β and its potential role in VaD, in particular, due to its multimodal effects in the brain and neuropathogenesis. ...
... Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73]. ...
... Strong evidence for the role of TGF-β1 in VaD is derived from studies using mice overexpressing TGF-β1. These mice have originally been generated by Tony Wyss-Coray and colleagues and are characterized by massive deposition of ECM and the formation of hydrocephalus [74]. This mouse model has a higher susceptibility to neuroinflammation triggered by experimental autoimmune encephalitis [75], accelerated disease progression in the context of amyotrophic lateral sclerosis (ALS) [76], severe formation of amyloid plaque pathology in the context of an AD background [69], microvascular degenerations [62], reduced level of neurogenesis [77], and most importantly, learning deficits [78]. More specifically in the context of VaD, these mice show reduced functional vascular reactivity in the sense of a lower dilatation as well as contraction behavior in response to stimuli, capillary degenerations resulting in the formation of string vessels, vascular fibrosis and calcification [79]. Importantly, in the scenario of VaD, the TGF-β1 overexpressing mice show reduced CBF, which correlates with the deposition of amyloid plaques in the blood vessels [80]. ...
A TGF-beta1 polymorphism association with dementia and neuropathologies: the HAAS
1
2007
... Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73]. ...
Genetics of Vascular Dementia: Systematic Review and Meta-Analysis
1
2015
... Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73]. ...
Chitotriosidase and inflammatory mediator levels in Alzheimer's disease and cerebrovascular dementia
1
2006
... Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73]. ...
Cerebrovascular transforming growth factor-beta contributes to inflammation in the Alzheimer's disease brain
1
2002
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Interleukin-18 and transforming growth factor-beta 1 plasma levels in Alzheimer's disease and vascular dementia
0
2006
An endothelial link between the benefits of physical exercise in dementia
0
2017
Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer's disease
2
1997
... Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73]. ...
... Strong evidence for the role of TGF-β1 in VaD is derived from studies using mice overexpressing TGF-β1. These mice have originally been generated by Tony Wyss-Coray and colleagues and are characterized by massive deposition of ECM and the formation of hydrocephalus [74]. This mouse model has a higher susceptibility to neuroinflammation triggered by experimental autoimmune encephalitis [75], accelerated disease progression in the context of amyotrophic lateral sclerosis (ALS) [76], severe formation of amyloid plaque pathology in the context of an AD background [69], microvascular degenerations [62], reduced level of neurogenesis [77], and most importantly, learning deficits [78]. More specifically in the context of VaD, these mice show reduced functional vascular reactivity in the sense of a lower dilatation as well as contraction behavior in response to stimuli, capillary degenerations resulting in the formation of string vessels, vascular fibrosis and calcification [79]. Importantly, in the scenario of VaD, the TGF-β1 overexpressing mice show reduced CBF, which correlates with the deposition of amyloid plaques in the blood vessels [80]. ...
Increased expression of TGF-beta 1 in brain tissue after ischemic stroke in humans
2
1996
... Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73]. ...
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
Circulating transforming growth factor beta-1: a partial molecular explanation for associations between hypertension, diabetes, obesity, smoking and human disease involving fibrosis
1
2005
... Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73]. ...
Cerebral small vessel disease-related protease HtrA1 processes latent TGF-beta binding protein 1 and facilitates TGF-beta signaling
1
2014
... Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73]. ...
Review: molecular genetics and pathology of hereditary small vessel diseases of the brain
1
2011
... Evidence accumulating from human genetic association studies, from expression analyses and animal experiments strongly suggest that TGF-β plays a role in the pathogenesis of VaD. For example, a meta-analysis of 69 studies with 4,462 cases and 11,583 controls identified polymorphisms in the TGF-β1 gene to be associated with a higher risk for VaD, microinfarcts, ischemic stroke and cerebral amyloid angiopathy [34, 63, 64]. Also, TGF-β1 levels are increased in monocyte-derived macrophages, plasma, in the cerebrovascular system, as well as in the cerebrospinal fluid in patients and animal models of VaD [60, 62, 65-69]. and in hypertensive, diabetic and ischemic stroke patients [70, 71]. In CARASIL, a hereditary small vessel disease, mutations in the serine protease HTRA1 gene result in increased TGF-β signaling, provoking multiple effects including vascular fibrosis and extracellular matrix synthesis [72, 73]. ...
Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-beta 1
3
1995
... Strong evidence for the role of TGF-β1 in VaD is derived from studies using mice overexpressing TGF-β1. These mice have originally been generated by Tony Wyss-Coray and colleagues and are characterized by massive deposition of ECM and the formation of hydrocephalus [74]. This mouse model has a higher susceptibility to neuroinflammation triggered by experimental autoimmune encephalitis [75], accelerated disease progression in the context of amyotrophic lateral sclerosis (ALS) [76], severe formation of amyloid plaque pathology in the context of an AD background [69], microvascular degenerations [62], reduced level of neurogenesis [77], and most importantly, learning deficits [78]. More specifically in the context of VaD, these mice show reduced functional vascular reactivity in the sense of a lower dilatation as well as contraction behavior in response to stimuli, capillary degenerations resulting in the formation of string vessels, vascular fibrosis and calcification [79]. Importantly, in the scenario of VaD, the TGF-β1 overexpressing mice show reduced CBF, which correlates with the deposition of amyloid plaques in the blood vessels [80]. ...
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
... Besides monocytes and macrophages, astrocytes are involved in the pathology of VaD. For example, astrogliosis is an acute (7 days) and chronic (3 months) response to artery occlusion in rats [181-183]. Here, in contrast to the effects on microglia, TGF-β seems to have detrimental effects. For example, TGF-β induces astrogliosis and is the main driver of gliotic scarring [74]. In the context of VaD, TGF-β overexpression induces a thickened vascular wall due to accumulation of extracellular matrix proteins [184]. In summary, TGF-β plays a crucial role in neuroinflammation, in particular, in the context of microglial and astroglial cells. ...
Astroglial overproduction of TGF-beta 1 enhances inflammatory central nervous system disease in transgenic mice
1
1997
... Strong evidence for the role of TGF-β1 in VaD is derived from studies using mice overexpressing TGF-β1. These mice have originally been generated by Tony Wyss-Coray and colleagues and are characterized by massive deposition of ECM and the formation of hydrocephalus [74]. This mouse model has a higher susceptibility to neuroinflammation triggered by experimental autoimmune encephalitis [75], accelerated disease progression in the context of amyotrophic lateral sclerosis (ALS) [76], severe formation of amyloid plaque pathology in the context of an AD background [69], microvascular degenerations [62], reduced level of neurogenesis [77], and most importantly, learning deficits [78]. More specifically in the context of VaD, these mice show reduced functional vascular reactivity in the sense of a lower dilatation as well as contraction behavior in response to stimuli, capillary degenerations resulting in the formation of string vessels, vascular fibrosis and calcification [79]. Importantly, in the scenario of VaD, the TGF-β1 overexpressing mice show reduced CBF, which correlates with the deposition of amyloid plaques in the blood vessels [80]. ...
Astrocyte-derived TGF-beta1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells
2
2015
... Strong evidence for the role of TGF-β1 in VaD is derived from studies using mice overexpressing TGF-β1. These mice have originally been generated by Tony Wyss-Coray and colleagues and are characterized by massive deposition of ECM and the formation of hydrocephalus [74]. This mouse model has a higher susceptibility to neuroinflammation triggered by experimental autoimmune encephalitis [75], accelerated disease progression in the context of amyotrophic lateral sclerosis (ALS) [76], severe formation of amyloid plaque pathology in the context of an AD background [69], microvascular degenerations [62], reduced level of neurogenesis [77], and most importantly, learning deficits [78]. More specifically in the context of VaD, these mice show reduced functional vascular reactivity in the sense of a lower dilatation as well as contraction behavior in response to stimuli, capillary degenerations resulting in the formation of string vessels, vascular fibrosis and calcification [79]. Importantly, in the scenario of VaD, the TGF-β1 overexpressing mice show reduced CBF, which correlates with the deposition of amyloid plaques in the blood vessels [80]. ...
... Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... Strong evidence for the role of TGF-β1 in VaD is derived from studies using mice overexpressing TGF-β1. These mice have originally been generated by Tony Wyss-Coray and colleagues and are characterized by massive deposition of ECM and the formation of hydrocephalus [74]. This mouse model has a higher susceptibility to neuroinflammation triggered by experimental autoimmune encephalitis [75], accelerated disease progression in the context of amyotrophic lateral sclerosis (ALS) [76], severe formation of amyloid plaque pathology in the context of an AD background [69], microvascular degenerations [62], reduced level of neurogenesis [77], and most importantly, learning deficits [78]. More specifically in the context of VaD, these mice show reduced functional vascular reactivity in the sense of a lower dilatation as well as contraction behavior in response to stimuli, capillary degenerations resulting in the formation of string vessels, vascular fibrosis and calcification [79]. Importantly, in the scenario of VaD, the TGF-β1 overexpressing mice show reduced CBF, which correlates with the deposition of amyloid plaques in the blood vessels [80]. ...
... Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
Chronic over-expression of TGFbeta1 alters hippocampal structure and causes learning deficits
1
2013
... Strong evidence for the role of TGF-β1 in VaD is derived from studies using mice overexpressing TGF-β1. These mice have originally been generated by Tony Wyss-Coray and colleagues and are characterized by massive deposition of ECM and the formation of hydrocephalus [74]. This mouse model has a higher susceptibility to neuroinflammation triggered by experimental autoimmune encephalitis [75], accelerated disease progression in the context of amyotrophic lateral sclerosis (ALS) [76], severe formation of amyloid plaque pathology in the context of an AD background [69], microvascular degenerations [62], reduced level of neurogenesis [77], and most importantly, learning deficits [78]. More specifically in the context of VaD, these mice show reduced functional vascular reactivity in the sense of a lower dilatation as well as contraction behavior in response to stimuli, capillary degenerations resulting in the formation of string vessels, vascular fibrosis and calcification [79]. Importantly, in the scenario of VaD, the TGF-β1 overexpressing mice show reduced CBF, which correlates with the deposition of amyloid plaques in the blood vessels [80]. ...
Simvastatin restored vascular reactivity, endothelial function and reduced string vessel pathology in a mouse model of cerebrovascular disease
1
2015
... Strong evidence for the role of TGF-β1 in VaD is derived from studies using mice overexpressing TGF-β1. These mice have originally been generated by Tony Wyss-Coray and colleagues and are characterized by massive deposition of ECM and the formation of hydrocephalus [74]. This mouse model has a higher susceptibility to neuroinflammation triggered by experimental autoimmune encephalitis [75], accelerated disease progression in the context of amyotrophic lateral sclerosis (ALS) [76], severe formation of amyloid plaque pathology in the context of an AD background [69], microvascular degenerations [62], reduced level of neurogenesis [77], and most importantly, learning deficits [78]. More specifically in the context of VaD, these mice show reduced functional vascular reactivity in the sense of a lower dilatation as well as contraction behavior in response to stimuli, capillary degenerations resulting in the formation of string vessels, vascular fibrosis and calcification [79]. Importantly, in the scenario of VaD, the TGF-β1 overexpressing mice show reduced CBF, which correlates with the deposition of amyloid plaques in the blood vessels [80]. ...
... Strong evidence for the role of TGF-β1 in VaD is derived from studies using mice overexpressing TGF-β1. These mice have originally been generated by Tony Wyss-Coray and colleagues and are characterized by massive deposition of ECM and the formation of hydrocephalus [74]. This mouse model has a higher susceptibility to neuroinflammation triggered by experimental autoimmune encephalitis [75], accelerated disease progression in the context of amyotrophic lateral sclerosis (ALS) [76], severe formation of amyloid plaque pathology in the context of an AD background [69], microvascular degenerations [62], reduced level of neurogenesis [77], and most importantly, learning deficits [78]. More specifically in the context of VaD, these mice show reduced functional vascular reactivity in the sense of a lower dilatation as well as contraction behavior in response to stimuli, capillary degenerations resulting in the formation of string vessels, vascular fibrosis and calcification [79]. Importantly, in the scenario of VaD, the TGF-β1 overexpressing mice show reduced CBF, which correlates with the deposition of amyloid plaques in the blood vessels [80]. ...
Increased levels of transforming growth factor-beta1 in essential hypertension
1
2002
... Reduced CBF is the hallmark of VaD, which may be the result of very diverse conditions such as atherosclerosis, cerebral amyloid angiopathy, microbleeding, micro-strokes and larger territorial stroke. Such damages typically induce a rise in TGF-β1 levels, which locally affects neurodegenerative and neuroregenerative events. The causative role of TGF-β in priming and development of CBF and VaD is obscure. Nevertheless, in patients with hypertension, the main risk factor for VaD, plasma levels of TGF-β1 are markedly increased [81] and this correlates with hypertension-induced tissue and organ damage such as nephrosclerosis and kidney diseases [82]. Thus, the observed changes in CBF seen in neurovascular pathologies might be strongly associated with the increased levels of TGF-mediated defects in the cellular components of the BBB. ...
Transforming growth factor beta signaling, vascular remodeling, and hypertension
1
2006
... Reduced CBF is the hallmark of VaD, which may be the result of very diverse conditions such as atherosclerosis, cerebral amyloid angiopathy, microbleeding, micro-strokes and larger territorial stroke. Such damages typically induce a rise in TGF-β1 levels, which locally affects neurodegenerative and neuroregenerative events. The causative role of TGF-β in priming and development of CBF and VaD is obscure. Nevertheless, in patients with hypertension, the main risk factor for VaD, plasma levels of TGF-β1 are markedly increased [81] and this correlates with hypertension-induced tissue and organ damage such as nephrosclerosis and kidney diseases [82]. Thus, the observed changes in CBF seen in neurovascular pathologies might be strongly associated with the increased levels of TGF-mediated defects in the cellular components of the BBB. ...
Pericytes and Neurovascular Function in the Healthy and Diseased Brain
1
2019
... The BBB serves as a remarkable physical and biochemical barricade between the brain and the blood. The BBB is primarily built up by endothelial cells lining the microvessels and sealed through tight junctions. The vessel wall is ensheathed by pericytes and surrounded by end-feet of perivascular astrocytes, microglial cells, and neurons, called neurovascular unit (NVU) [83-87]. In the healthy brain, the BBB protects neurons from blood-derived factors and maintains levels of neurochemical parameters of the internal brain milieu, which are essential for neuroprotection, neuronal functioning and synaptic transmission [84, 88]. This specialized function of the BBB is maintained by the establishment of a mutual trophic interference and three-dimensional vicinity between endothelial cells, pericytes and astrocytes in the brain. ...
The blood-brain barrier
1
2015
... The BBB serves as a remarkable physical and biochemical barricade between the brain and the blood. The BBB is primarily built up by endothelial cells lining the microvessels and sealed through tight junctions. The vessel wall is ensheathed by pericytes and surrounded by end-feet of perivascular astrocytes, microglial cells, and neurons, called neurovascular unit (NVU) [83-87]. In the healthy brain, the BBB protects neurons from blood-derived factors and maintains levels of neurochemical parameters of the internal brain milieu, which are essential for neuroprotection, neuronal functioning and synaptic transmission [84, 88]. This specialized function of the BBB is maintained by the establishment of a mutual trophic interference and three-dimensional vicinity between endothelial cells, pericytes and astrocytes in the brain. ...
The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease
1
2017
... Neuroinflammation, like in many other neuro-degenerative diseases, is one of the crucial hallmarks of VaD [146]. It describes a wide range of inflammatory responses within the CNS and is mediated by the production of cytokines, chemokines, and reactive oxygen species. At the cellular level, neuroinflammation involves mainly myeloid cells, i.e. microglia, monocytes, and macrophages, and other peripherally derived immune cells infiltrating the CNS, as well as astrocytes. In response to vascular injuries, microglia are the first cells to respond. Initially classified as M1 pro-inflammatory and anti-regenerative, and M2 anti-inflammatory and pro-regenerative, it is now clear that microglia appear in many different subtypes and phenotypes, with high plasticity and transition between the various types [169-171]. Though microglia have altered activity in VaD even before clinical symptoms and neuropathological hallmarks arise [85, 172], their precise role in VaD is unclear. Nevertheless, under inflammatory conditions, microglia are producers of IL-1b [173], and higher expression of IL-1b has been reported in the brain of hypertensive rats [174]. Also, microglia may contribute to CBF alterations and hypertensive vascular pathology in the brain [175]. Besides microglia, perivascular macrophages might be involved in particular. Monocytes are attracted to the vascular wall by vascular damage, where they differentiate into macrophages, locally secrete matrix proteins, vasculo-protective and pro-angiogenic factors as well as several other growth factors [176], promoting angiogenesis and functional recovery [177]. ...
The neurovascular unit - concept review
0
2014
Development, maintenance and disruption of the blood-brain barrier
1
2013
... The BBB serves as a remarkable physical and biochemical barricade between the brain and the blood. The BBB is primarily built up by endothelial cells lining the microvessels and sealed through tight junctions. The vessel wall is ensheathed by pericytes and surrounded by end-feet of perivascular astrocytes, microglial cells, and neurons, called neurovascular unit (NVU) [83-87]. In the healthy brain, the BBB protects neurons from blood-derived factors and maintains levels of neurochemical parameters of the internal brain milieu, which are essential for neuroprotection, neuronal functioning and synaptic transmission [84, 88]. This specialized function of the BBB is maintained by the establishment of a mutual trophic interference and three-dimensional vicinity between endothelial cells, pericytes and astrocytes in the brain. ...
The blood-brain barrier: an overview: structure, regulation, and clinical implications
1
2004
... The BBB serves as a remarkable physical and biochemical barricade between the brain and the blood. The BBB is primarily built up by endothelial cells lining the microvessels and sealed through tight junctions. The vessel wall is ensheathed by pericytes and surrounded by end-feet of perivascular astrocytes, microglial cells, and neurons, called neurovascular unit (NVU) [83-87]. In the healthy brain, the BBB protects neurons from blood-derived factors and maintains levels of neurochemical parameters of the internal brain milieu, which are essential for neuroprotection, neuronal functioning and synaptic transmission [84, 88]. This specialized function of the BBB is maintained by the establishment of a mutual trophic interference and three-dimensional vicinity between endothelial cells, pericytes and astrocytes in the brain. ...
Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke
1
2011
... The potential relationship between TGF-β and the BBB is demonstrated in gene knockout studies as complete deletion of TGF-β1, TGF-βR1, TGF-βR2, TGF-βR3 (endoglin) and of Smad4. Those resulted in defects in blood vessel formation and embryonic lethality due to an improper attachment between endothelial and mesothelial cells. Systemic inhibition of TGF-β by soluble endoglin in pregnant rats contributes to the development of Preeclampsia, a disease condition associated with increased BBB leakage in pregnant women. Moreover, a Cre-LoxP based conditional deletion of Smad-4 specifically in brain endothelial cells resulted in BBB breakdown due to disruption in the interaction between pericytes and endothelial cells. Besides, TGF-β contributes to the construction of basement membrane through the secretion of ECM components in endothelial cells and pericytes that are integral parts of the BBB [89-91]. Moreover, the increased autocrine TGF-β signaling in pericytes in diabetic encephalopathy induces basement membrane hypertrophy disrupting the BBB [92]. Taken together, TGF-β plays a key role in the recruitment of pericytes and maintenance of the contact between endothelial cells and pericytes at the BBB (Fig. 3). However, the TGF-β mediated recruitment of astrocytes to the BBB and the complete molecular basis of interactions among cellular components of the BBB remain largely unknown. ...
Formation and maintenance of the BBB
0
2015
Diverse cell signaling events modulated by perlecan
1
2008
... The potential relationship between TGF-β and the BBB is demonstrated in gene knockout studies as complete deletion of TGF-β1, TGF-βR1, TGF-βR2, TGF-βR3 (endoglin) and of Smad4. Those resulted in defects in blood vessel formation and embryonic lethality due to an improper attachment between endothelial and mesothelial cells. Systemic inhibition of TGF-β by soluble endoglin in pregnant rats contributes to the development of Preeclampsia, a disease condition associated with increased BBB leakage in pregnant women. Moreover, a Cre-LoxP based conditional deletion of Smad-4 specifically in brain endothelial cells resulted in BBB breakdown due to disruption in the interaction between pericytes and endothelial cells. Besides, TGF-β contributes to the construction of basement membrane through the secretion of ECM components in endothelial cells and pericytes that are integral parts of the BBB [89-91]. Moreover, the increased autocrine TGF-β signaling in pericytes in diabetic encephalopathy induces basement membrane hypertrophy disrupting the BBB [92]. Taken together, TGF-β plays a key role in the recruitment of pericytes and maintenance of the contact between endothelial cells and pericytes at the BBB (Fig. 3). However, the TGF-β mediated recruitment of astrocytes to the BBB and the complete molecular basis of interactions among cellular components of the BBB remain largely unknown. ...
Advanced glycation end-products disrupt the blood-brain barrier by stimulating the release of transforming growth factor-beta by pericytes and vascular endothelial growth factor and matrix metalloproteinase-2 by endothelial cells in vitro
1
2013
... The potential relationship between TGF-β and the BBB is demonstrated in gene knockout studies as complete deletion of TGF-β1, TGF-βR1, TGF-βR2, TGF-βR3 (endoglin) and of Smad4. Those resulted in defects in blood vessel formation and embryonic lethality due to an improper attachment between endothelial and mesothelial cells. Systemic inhibition of TGF-β by soluble endoglin in pregnant rats contributes to the development of Preeclampsia, a disease condition associated with increased BBB leakage in pregnant women. Moreover, a Cre-LoxP based conditional deletion of Smad-4 specifically in brain endothelial cells resulted in BBB breakdown due to disruption in the interaction between pericytes and endothelial cells. Besides, TGF-β contributes to the construction of basement membrane through the secretion of ECM components in endothelial cells and pericytes that are integral parts of the BBB [89-91]. Moreover, the increased autocrine TGF-β signaling in pericytes in diabetic encephalopathy induces basement membrane hypertrophy disrupting the BBB [92]. Taken together, TGF-β plays a key role in the recruitment of pericytes and maintenance of the contact between endothelial cells and pericytes at the BBB (Fig. 3). However, the TGF-β mediated recruitment of astrocytes to the BBB and the complete molecular basis of interactions among cellular components of the BBB remain largely unknown. ...
TGF-beta1 interactome: metastasis and beyond
1
2010
... In the context of cancer, increased TGF-β facilitates metastasis of tumor cells through the down-regulation of junctional adhesion and tight junction proteins upon the induction of matrix metalloproteases (MMPs) [35, 93]. Similarly, in neuroinflammatory conditions, down-regulation of the expression of junctional adhesion and tight junction proteins in the blood vessel and increased expression of MMPs in association with abnormal TGF-β signaling primes the leakage of BBB, which in turn leads to degeneration of endothelial cells, pericytes drifting and string vessels in the neurovascular systems [94]. Based on the observation from the ablation of the TGF-β signaling pathway in pericytes through retroviral-mediated expression of a truncated C-terminal intracellular kinase domain of TGF-βR2, Sieczkiewicz GJ et al. have described that endothelial cell-specific secretion of TGF-β inhibits the mitotic activities of pericytes through the activation of pericyte contractile protein leading to the BBB breakdown [95]. ...
Lack of Smad or Notch leads to a fatal game of brain pericyte hopscotch
1
2011
... In the context of cancer, increased TGF-β facilitates metastasis of tumor cells through the down-regulation of junctional adhesion and tight junction proteins upon the induction of matrix metalloproteases (MMPs) [35, 93]. Similarly, in neuroinflammatory conditions, down-regulation of the expression of junctional adhesion and tight junction proteins in the blood vessel and increased expression of MMPs in association with abnormal TGF-β signaling primes the leakage of BBB, which in turn leads to degeneration of endothelial cells, pericytes drifting and string vessels in the neurovascular systems [94]. Based on the observation from the ablation of the TGF-β signaling pathway in pericytes through retroviral-mediated expression of a truncated C-terminal intracellular kinase domain of TGF-βR2, Sieczkiewicz GJ et al. have described that endothelial cell-specific secretion of TGF-β inhibits the mitotic activities of pericytes through the activation of pericyte contractile protein leading to the BBB breakdown [95]. ...
TGF-beta 1 signaling controls retinal pericyte contractile protein expression
1
2003
... In the context of cancer, increased TGF-β facilitates metastasis of tumor cells through the down-regulation of junctional adhesion and tight junction proteins upon the induction of matrix metalloproteases (MMPs) [35, 93]. Similarly, in neuroinflammatory conditions, down-regulation of the expression of junctional adhesion and tight junction proteins in the blood vessel and increased expression of MMPs in association with abnormal TGF-β signaling primes the leakage of BBB, which in turn leads to degeneration of endothelial cells, pericytes drifting and string vessels in the neurovascular systems [94]. Based on the observation from the ablation of the TGF-β signaling pathway in pericytes through retroviral-mediated expression of a truncated C-terminal intracellular kinase domain of TGF-βR2, Sieczkiewicz GJ et al. have described that endothelial cell-specific secretion of TGF-β inhibits the mitotic activities of pericytes through the activation of pericyte contractile protein leading to the BBB breakdown [95]. ...
Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch
1
2011
... In a preclinical genetic mouse model of VaD with a point mutation in the NOTCH3 gene, TGF-β signaling is disrupted, and pericyte numbers are reduced leading to BBB breakdown. Notably, endothelial-specific Smad4 deficient animals had increased BBB permeability, BBB breakdown and intracranial haemorrhages [96]. Moreover, the animal models with endothelial cell-specific depletion of TGF-receptors including endoglin have cerebrovascular defects and BBB dysfunctions [97-101]. In humans, mutations in the TGF-β receptors have been linked to hereditary haemorrhagic telangiectasia (HHT). HHT is an autosomal dominant vascular dysplasia, caused by mutations in endoglin and ALK1, which is characterized by dilated vessels and arteriovenous malformations [102]. Eventually, the transgenic model of HHT is characterized by significant structural alterations in cerebral blood vessels, pericyte drift and BBB breakdown [103]. However, the role of TGF-β in the BBB breakdown and vessel leakage in VaD is currently not clear. While many cerebrovascular pathologies, including VaD, have been characterized by the increased level of TGF-β, defects in its downstream pathways and impact of the crosstalk between abnormal levels of TGF-β and other signaling mechanism needs to be considered. Future studies directed towards the potential involvement of TGF-β in BBB dysfunction would signify the therapeutic options for VaD. ...
Compensatory signalling induced in the yolk sac vasculature by deletion of TGFbeta receptors in mice
1
2007
... In a preclinical genetic mouse model of VaD with a point mutation in the NOTCH3 gene, TGF-β signaling is disrupted, and pericyte numbers are reduced leading to BBB breakdown. Notably, endothelial-specific Smad4 deficient animals had increased BBB permeability, BBB breakdown and intracranial haemorrhages [96]. Moreover, the animal models with endothelial cell-specific depletion of TGF-receptors including endoglin have cerebrovascular defects and BBB dysfunctions [97-101]. In humans, mutations in the TGF-β receptors have been linked to hereditary haemorrhagic telangiectasia (HHT). HHT is an autosomal dominant vascular dysplasia, caused by mutations in endoglin and ALK1, which is characterized by dilated vessels and arteriovenous malformations [102]. Eventually, the transgenic model of HHT is characterized by significant structural alterations in cerebral blood vessels, pericyte drift and BBB breakdown [103]. However, the role of TGF-β in the BBB breakdown and vessel leakage in VaD is currently not clear. While many cerebrovascular pathologies, including VaD, have been characterized by the increased level of TGF-β, defects in its downstream pathways and impact of the crosstalk between abnormal levels of TGF-β and other signaling mechanism needs to be considered. Future studies directed towards the potential involvement of TGF-β in BBB dysfunction would signify the therapeutic options for VaD. ...
Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models--brief report
0
2014
Pathogenesis of arteriovenous malformations in the absence of endoglin
0
2010
Angiogenesis regulation by TGFbeta signalling: clues from an inherited vascular disease
0
2011
Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression
1
2014
... In a preclinical genetic mouse model of VaD with a point mutation in the NOTCH3 gene, TGF-β signaling is disrupted, and pericyte numbers are reduced leading to BBB breakdown. Notably, endothelial-specific Smad4 deficient animals had increased BBB permeability, BBB breakdown and intracranial haemorrhages [96]. Moreover, the animal models with endothelial cell-specific depletion of TGF-receptors including endoglin have cerebrovascular defects and BBB dysfunctions [97-101]. In humans, mutations in the TGF-β receptors have been linked to hereditary haemorrhagic telangiectasia (HHT). HHT is an autosomal dominant vascular dysplasia, caused by mutations in endoglin and ALK1, which is characterized by dilated vessels and arteriovenous malformations [102]. Eventually, the transgenic model of HHT is characterized by significant structural alterations in cerebral blood vessels, pericyte drift and BBB breakdown [103]. However, the role of TGF-β in the BBB breakdown and vessel leakage in VaD is currently not clear. While many cerebrovascular pathologies, including VaD, have been characterized by the increased level of TGF-β, defects in its downstream pathways and impact of the crosstalk between abnormal levels of TGF-β and other signaling mechanism needs to be considered. Future studies directed towards the potential involvement of TGF-β in BBB dysfunction would signify the therapeutic options for VaD. ...
Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway
1
2006
... In a preclinical genetic mouse model of VaD with a point mutation in the NOTCH3 gene, TGF-β signaling is disrupted, and pericyte numbers are reduced leading to BBB breakdown. Notably, endothelial-specific Smad4 deficient animals had increased BBB permeability, BBB breakdown and intracranial haemorrhages [96]. Moreover, the animal models with endothelial cell-specific depletion of TGF-receptors including endoglin have cerebrovascular defects and BBB dysfunctions [97-101]. In humans, mutations in the TGF-β receptors have been linked to hereditary haemorrhagic telangiectasia (HHT). HHT is an autosomal dominant vascular dysplasia, caused by mutations in endoglin and ALK1, which is characterized by dilated vessels and arteriovenous malformations [102]. Eventually, the transgenic model of HHT is characterized by significant structural alterations in cerebral blood vessels, pericyte drift and BBB breakdown [103]. However, the role of TGF-β in the BBB breakdown and vessel leakage in VaD is currently not clear. While many cerebrovascular pathologies, including VaD, have been characterized by the increased level of TGF-β, defects in its downstream pathways and impact of the crosstalk between abnormal levels of TGF-β and other signaling mechanism needs to be considered. Future studies directed towards the potential involvement of TGF-β in BBB dysfunction would signify the therapeutic options for VaD. ...
Pericytes as targets in hereditary hemorrhagic telangiectasia
1
2015
... In a preclinical genetic mouse model of VaD with a point mutation in the NOTCH3 gene, TGF-β signaling is disrupted, and pericyte numbers are reduced leading to BBB breakdown. Notably, endothelial-specific Smad4 deficient animals had increased BBB permeability, BBB breakdown and intracranial haemorrhages [96]. Moreover, the animal models with endothelial cell-specific depletion of TGF-receptors including endoglin have cerebrovascular defects and BBB dysfunctions [97-101]. In humans, mutations in the TGF-β receptors have been linked to hereditary haemorrhagic telangiectasia (HHT). HHT is an autosomal dominant vascular dysplasia, caused by mutations in endoglin and ALK1, which is characterized by dilated vessels and arteriovenous malformations [102]. Eventually, the transgenic model of HHT is characterized by significant structural alterations in cerebral blood vessels, pericyte drift and BBB breakdown [103]. However, the role of TGF-β in the BBB breakdown and vessel leakage in VaD is currently not clear. While many cerebrovascular pathologies, including VaD, have been characterized by the increased level of TGF-β, defects in its downstream pathways and impact of the crosstalk between abnormal levels of TGF-β and other signaling mechanism needs to be considered. Future studies directed towards the potential involvement of TGF-β in BBB dysfunction would signify the therapeutic options for VaD. ...
The emerging concept of vascular remodeling
1
1994
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Brain arterial remodeling contribution to nonembolic brain infarcts in patients with HIV
0
2015
Vascular remodeling
0
1996
Pathophysiology of vascular remodeling in hypertension
1
2013
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Mechanisms of arterial remodeling: lessons from genetic diseases
1
2012
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis
2
2001
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
... ]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Vascular remodeling in hypertension: mechanisms and treatment
1
2012
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Cellular and molecular biology of vascular remodeling
1
1996
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Endothelial cells in physiology and in the pathophysiology of vascular disorders
1
1998
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Human vascular endothelial cells: a model system for studying vascular inflammation in diabetes and atherosclerosis
0
2011
The vascular endothelium and human diseases
1
2013
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Hypertension-induced organ damage in African Americans: transforming growth factor-beta(1) excess as a mechanism for increased prevalence
1
2000
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Transforming growth factor-beta-dependent events in vascular remodeling following arterial injury
1
2003
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Valvular endothelial cells and the mechanoregulation of valvular pathology
1
2007
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Endothelial cells respond to the direction of mechanical stimuli through SMAD signaling to regulate coronary artery size
1
2017
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Endothelial cells provide feedback control for vascular remodeling through a mechanosensitive autocrine TGF-beta signaling pathway
1
2008
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice
1
1995
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Deletion of Endothelial Transforming Growth Factor-beta Signaling Leads to Choroidal Neovascularization
1
2017
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
TGF-beta-induced apoptosis in endothelial cells mediated by M6P/IGFII-R and mini-plasminogen
1
2005
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Vascular cell apoptosis: cell type-specific modulation by transforming growth factor-beta1 in endothelial cells versus smooth muscle cells
1
1999
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Transforming growth factor-beta 1 modulates basic fibroblast growth factor-induced proteolytic and angiogenic properties of endothelial cells in vitro
1
1990
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
The opposing effects of basic fibroblast growth factor and transforming growth factor beta on the regulation of plasminogen activator activity in capillary endothelial cells
1
1987
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
TGF-beta1 induces endothelial cell apoptosis by shifting VEGF activation of p38(MAPK) from the prosurvival p38beta to proapoptotic p38alpha
2
2012
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
... ]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
The response of endothelial cells to TGF beta-1 is dependent upon cell shape, proliferative state and the nature of the substratum
1
1991
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Cerebrovascular endothelial dysfunction mediated by beta-amyloid
1
1997
... Vascular remodeling (VR) is a dynamic process of structural plasticity of blood vessels in which the blood vessel cells are constantly altered at the level of cell growth, cell migration, cell death, and at the level of production and degradation of ECM components present in the basement membrane [104-108]. This can be in response to variation in blood flow and hypertension, and also as a consequence of lesions [107, 109, 110]. Interactions between vascular cells mediate the regulation of VR through signaling of growth factors, vasoactive substances and hemodynamic stimuli [109, 111]. Endothelial cells contribute to vascular hemostasis, interact with circulating blood cells, provide resistance over the hemodynamic tension and mechanical stimuli from blood pressure, and respond to inflammation [112-114]. TGF-β signaling is one of the key pathways involved in the regulation of VR [115, 116]. In general, mechanical stimuli are crucial regulators of growth as well as degeneration of endothelial cells that contribute to VR [117, 118]. Spontaneously hypertensive rats with altered hemodynamics exhibited increased levels of endothelial heparan sulfate proteoglycan (HSPG), another key component of basement membrane regulated by TGF-β-Smad signaling [119]. While TGF-β1 deficient mice are embryonic lethal due to defects in vasculogenesis [120], in vivo induction of TGF-β1 appears to mediate angiogenesis. TGF-β has been reported to exert differential effects on vascular endothelial cells. In general, the formation of new capillaries from preexisting blood vessels is mediated by TGF-β1 induced expression of endothelial cell VEGF through fibroblast growth factor (FGF-2)[47]. Recently, Schlecht et al. described that TGF-β is required for physiological vasculogenesis in the eye, as deletion of TGF-βR2 especially in endothelial cells of the eye results in choroidal neovascularization [121]. Moreover, in vitro experiments of cultured endothelial cells suggest that TGF-β inhibits the mitotic activity and induces cell death of endothelial cells [122, 123]. In addition, prolonged treatment of vascular endothelial cells together with TGF-β and FGF-2 abolishes the mitogenic effect of FGF-2 on endothelial cell function and migration through the deactivation of plasminogen activator [124, 125]. Apparently, there exists a positive feedback loop from TGF-β1 induced VEGF expression that in turn boosts the expression of TGF-β, which results in the activation of p38 mitogen-activated protein kinase (MAPK) leading to the degeneration of endothelial cells during chronic neuroinflammation and vascular pathogenesis [126]. When endothelial cell death occurs, the basement membrane persists as empty tubes known as string or ghost vessels. Such TGF-β induced degeneration in endothelial cells appears to induce string vessels in the brain [126], which are often associated with neurovascular pathogenesis in AD and VaD [59]. Microvessels isolated from brains of AD subjects with vascular pathologies are also reported to release TGF-β [66]. In the context of cerebral amyloid angiopathy, TGF-β appears to induce the generation of amyloid-β depositions in blood vessels. Thus, TGF-β mediated accumulation of amyloid-β might also be closely associated with the endothelial cell death leading to cerebrovascular pathologies in VaD [127, 128]. ...
Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology
2
2019
... Recently, endothelial to mesenchymal transition (EndoMT) has been proposed as a potential mechanism involved in VR and BBB breakdown in cerebrovascular pathologies [129, 130]. EndoMT is a cellular process by which endothelial cells dedifferentiate into mesenchymal cells leading to degradation of vascular basement membrane and disintegration of endothelial cells [131]. Moreover, physiological properties and functions of endothelial cells are impaired, and they acquire a migratory phenotype. Notably, cardiac fibrosis, metastatic conditions, systemic sclerosis, multiple sclerosis, stroke, cerebral cavernous malformation, vein stenosis and diabetic retinopathy have been characterized by EndoMT [132-134]. Troletti et al. demonstrated that neuro-inflammation-mediated TGF-β and IL-1 β play a key role in EndoMT in the vasculature of human post-mortem brains with multiple sclerosis and cerebrovascular abnormalities [129]. Moreover, inhibition of TGF-β signaling by neutralizing antibodies, endothelial cell-specific Smad2/3 deletion, shRNA-mediated knockdown of Smad2 and haploinsufficiency of Smad3 minimize EndoMT [135, 136].Therefore, abnormal circulation of TGF-β might contribute to VR and breakdown of BBB through EndoMT in VaD. ...
... ]. Troletti et al. demonstrated that neuro-inflammation-mediated TGF-β and IL-1 β play a key role in EndoMT in the vasculature of human post-mortem brains with multiple sclerosis and cerebrovascular abnormalities [129]. Moreover, inhibition of TGF-β signaling by neutralizing antibodies, endothelial cell-specific Smad2/3 deletion, shRNA-mediated knockdown of Smad2 and haploinsufficiency of Smad3 minimize EndoMT [135, 136].Therefore, abnormal circulation of TGF-β might contribute to VR and breakdown of BBB through EndoMT in VaD. ...
Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review
1
2019
... Recently, endothelial to mesenchymal transition (EndoMT) has been proposed as a potential mechanism involved in VR and BBB breakdown in cerebrovascular pathologies [129, 130]. EndoMT is a cellular process by which endothelial cells dedifferentiate into mesenchymal cells leading to degradation of vascular basement membrane and disintegration of endothelial cells [131]. Moreover, physiological properties and functions of endothelial cells are impaired, and they acquire a migratory phenotype. Notably, cardiac fibrosis, metastatic conditions, systemic sclerosis, multiple sclerosis, stroke, cerebral cavernous malformation, vein stenosis and diabetic retinopathy have been characterized by EndoMT [132-134]. Troletti et al. demonstrated that neuro-inflammation-mediated TGF-β and IL-1 β play a key role in EndoMT in the vasculature of human post-mortem brains with multiple sclerosis and cerebrovascular abnormalities [129]. Moreover, inhibition of TGF-β signaling by neutralizing antibodies, endothelial cell-specific Smad2/3 deletion, shRNA-mediated knockdown of Smad2 and haploinsufficiency of Smad3 minimize EndoMT [135, 136].Therefore, abnormal circulation of TGF-β might contribute to VR and breakdown of BBB through EndoMT in VaD. ...
Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders
1
2011
... Recently, endothelial to mesenchymal transition (EndoMT) has been proposed as a potential mechanism involved in VR and BBB breakdown in cerebrovascular pathologies [129, 130]. EndoMT is a cellular process by which endothelial cells dedifferentiate into mesenchymal cells leading to degradation of vascular basement membrane and disintegration of endothelial cells [131]. Moreover, physiological properties and functions of endothelial cells are impaired, and they acquire a migratory phenotype. Notably, cardiac fibrosis, metastatic conditions, systemic sclerosis, multiple sclerosis, stroke, cerebral cavernous malformation, vein stenosis and diabetic retinopathy have been characterized by EndoMT [132-134]. Troletti et al. demonstrated that neuro-inflammation-mediated TGF-β and IL-1 β play a key role in EndoMT in the vasculature of human post-mortem brains with multiple sclerosis and cerebrovascular abnormalities [129]. Moreover, inhibition of TGF-β signaling by neutralizing antibodies, endothelial cell-specific Smad2/3 deletion, shRNA-mediated knockdown of Smad2 and haploinsufficiency of Smad3 minimize EndoMT [135, 136].Therefore, abnormal circulation of TGF-β might contribute to VR and breakdown of BBB through EndoMT in VaD. ...
Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension
1
2015
... Recently, endothelial to mesenchymal transition (EndoMT) has been proposed as a potential mechanism involved in VR and BBB breakdown in cerebrovascular pathologies [129, 130]. EndoMT is a cellular process by which endothelial cells dedifferentiate into mesenchymal cells leading to degradation of vascular basement membrane and disintegration of endothelial cells [131]. Moreover, physiological properties and functions of endothelial cells are impaired, and they acquire a migratory phenotype. Notably, cardiac fibrosis, metastatic conditions, systemic sclerosis, multiple sclerosis, stroke, cerebral cavernous malformation, vein stenosis and diabetic retinopathy have been characterized by EndoMT [132-134]. Troletti et al. demonstrated that neuro-inflammation-mediated TGF-β and IL-1 β play a key role in EndoMT in the vasculature of human post-mortem brains with multiple sclerosis and cerebrovascular abnormalities [129]. Moreover, inhibition of TGF-β signaling by neutralizing antibodies, endothelial cell-specific Smad2/3 deletion, shRNA-mediated knockdown of Smad2 and haploinsufficiency of Smad3 minimize EndoMT [135, 136].Therefore, abnormal circulation of TGF-β might contribute to VR and breakdown of BBB through EndoMT in VaD. ...
Endothelial-mesenchymal transition of brain endothelial cells: possible role during metastatic extravasation
0
2015
The therapeutic potential of targeting the endothelial-to-mesenchymal transition
1
2019
... Recently, endothelial to mesenchymal transition (EndoMT) has been proposed as a potential mechanism involved in VR and BBB breakdown in cerebrovascular pathologies [129, 130]. EndoMT is a cellular process by which endothelial cells dedifferentiate into mesenchymal cells leading to degradation of vascular basement membrane and disintegration of endothelial cells [131]. Moreover, physiological properties and functions of endothelial cells are impaired, and they acquire a migratory phenotype. Notably, cardiac fibrosis, metastatic conditions, systemic sclerosis, multiple sclerosis, stroke, cerebral cavernous malformation, vein stenosis and diabetic retinopathy have been characterized by EndoMT [132-134]. Troletti et al. demonstrated that neuro-inflammation-mediated TGF-β and IL-1 β play a key role in EndoMT in the vasculature of human post-mortem brains with multiple sclerosis and cerebrovascular abnormalities [129]. Moreover, inhibition of TGF-β signaling by neutralizing antibodies, endothelial cell-specific Smad2/3 deletion, shRNA-mediated knockdown of Smad2 and haploinsufficiency of Smad3 minimize EndoMT [135, 136].Therefore, abnormal circulation of TGF-β might contribute to VR and breakdown of BBB through EndoMT in VaD. ...
TGF-beta signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling
1
2014
... Recently, endothelial to mesenchymal transition (EndoMT) has been proposed as a potential mechanism involved in VR and BBB breakdown in cerebrovascular pathologies [129, 130]. EndoMT is a cellular process by which endothelial cells dedifferentiate into mesenchymal cells leading to degradation of vascular basement membrane and disintegration of endothelial cells [131]. Moreover, physiological properties and functions of endothelial cells are impaired, and they acquire a migratory phenotype. Notably, cardiac fibrosis, metastatic conditions, systemic sclerosis, multiple sclerosis, stroke, cerebral cavernous malformation, vein stenosis and diabetic retinopathy have been characterized by EndoMT [132-134]. Troletti et al. demonstrated that neuro-inflammation-mediated TGF-β and IL-1 β play a key role in EndoMT in the vasculature of human post-mortem brains with multiple sclerosis and cerebrovascular abnormalities [129]. Moreover, inhibition of TGF-β signaling by neutralizing antibodies, endothelial cell-specific Smad2/3 deletion, shRNA-mediated knockdown of Smad2 and haploinsufficiency of Smad3 minimize EndoMT [135, 136].Therefore, abnormal circulation of TGF-β might contribute to VR and breakdown of BBB through EndoMT in VaD. ...
TGF-beta-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases
1
2017
... Recently, endothelial to mesenchymal transition (EndoMT) has been proposed as a potential mechanism involved in VR and BBB breakdown in cerebrovascular pathologies [129, 130]. EndoMT is a cellular process by which endothelial cells dedifferentiate into mesenchymal cells leading to degradation of vascular basement membrane and disintegration of endothelial cells [131]. Moreover, physiological properties and functions of endothelial cells are impaired, and they acquire a migratory phenotype. Notably, cardiac fibrosis, metastatic conditions, systemic sclerosis, multiple sclerosis, stroke, cerebral cavernous malformation, vein stenosis and diabetic retinopathy have been characterized by EndoMT [132-134]. Troletti et al. demonstrated that neuro-inflammation-mediated TGF-β and IL-1 β play a key role in EndoMT in the vasculature of human post-mortem brains with multiple sclerosis and cerebrovascular abnormalities [129]. Moreover, inhibition of TGF-β signaling by neutralizing antibodies, endothelial cell-specific Smad2/3 deletion, shRNA-mediated knockdown of Smad2 and haploinsufficiency of Smad3 minimize EndoMT [135, 136].Therefore, abnormal circulation of TGF-β might contribute to VR and breakdown of BBB through EndoMT in VaD. ...
Cellular mechanisms of CNS pericytes
2
2000
... Pericytes are multifunctional cells which ensheath the blood vessel and contribute to the regulation of capillary diameter, vasoconstriction and vasodilation, capillary blood flow and extracellular matrix protein secretion [33, 137, 138]. Moreover, pericytes have an immune-modulatory role in the brain [139]. With reference to the origin of the pericytes, both the mesenchymal and neural crest cells have been proposed to give rise to pericytes in many organs. It has recently been reported that pericytes play important roles in the homeostasis of the brain [140-142]. Pericytes are positioned within the BBB between endothelial cells, astrocytes and neurons [138]. Thus, pericytes can integrate and process signals from their adjacent cells to mediate diverse functional responses that are important for brain function in health and disease, including the regulation of the BBB permeability, angiogenesis, clearance of toxic metabolites, phagocytotic activity, capillary hemodynamic responses and neuroinflammation. ...
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
Pericytes of the neurovascular unit: key functions and signaling pathways
3
2016
... Pericytes are multifunctional cells which ensheath the blood vessel and contribute to the regulation of capillary diameter, vasoconstriction and vasodilation, capillary blood flow and extracellular matrix protein secretion [33, 137, 138]. Moreover, pericytes have an immune-modulatory role in the brain [139]. With reference to the origin of the pericytes, both the mesenchymal and neural crest cells have been proposed to give rise to pericytes in many organs. It has recently been reported that pericytes play important roles in the homeostasis of the brain [140-142]. Pericytes are positioned within the BBB between endothelial cells, astrocytes and neurons [138]. Thus, pericytes can integrate and process signals from their adjacent cells to mediate diverse functional responses that are important for brain function in health and disease, including the regulation of the BBB permeability, angiogenesis, clearance of toxic metabolites, phagocytotic activity, capillary hemodynamic responses and neuroinflammation. ...
... ]. Pericytes are positioned within the BBB between endothelial cells, astrocytes and neurons [138]. Thus, pericytes can integrate and process signals from their adjacent cells to mediate diverse functional responses that are important for brain function in health and disease, including the regulation of the BBB permeability, angiogenesis, clearance of toxic metabolites, phagocytotic activity, capillary hemodynamic responses and neuroinflammation. ...
... In human post-mortem AD brains and transgenic animal models of AD, pericytes are dysfunctional and they undergo degeneration [143-145]. Also, levels of soluble platelet-derived growth factor receptor β (PDGFR-β), a biomarker for pericyte death, are increased in cerebrospinal fluid (CSF) of subjects with mild dementia and in transgenic AD animals [138, 146, 147]. Thus, there is evidence for pericytes being involved in VaD, although the underlying mechanisms are largely unknown. Nevertheless, a familial form of VaD, CADASIL is associated with dysfunction of pericytes in the brain [148]. ...
Immune Regulation by Pericytes: Modulating Innate and Adaptive Immunity
1
2016
... Pericytes are multifunctional cells which ensheath the blood vessel and contribute to the regulation of capillary diameter, vasoconstriction and vasodilation, capillary blood flow and extracellular matrix protein secretion [33, 137, 138]. Moreover, pericytes have an immune-modulatory role in the brain [139]. With reference to the origin of the pericytes, both the mesenchymal and neural crest cells have been proposed to give rise to pericytes in many organs. It has recently been reported that pericytes play important roles in the homeostasis of the brain [140-142]. Pericytes are positioned within the BBB between endothelial cells, astrocytes and neurons [138]. Thus, pericytes can integrate and process signals from their adjacent cells to mediate diverse functional responses that are important for brain function in health and disease, including the regulation of the BBB permeability, angiogenesis, clearance of toxic metabolites, phagocytotic activity, capillary hemodynamic responses and neuroinflammation. ...
Clarification of mural cell coverage of vascular endothelial cells by live imaging of zebrafish
1
2016
... Pericytes are multifunctional cells which ensheath the blood vessel and contribute to the regulation of capillary diameter, vasoconstriction and vasodilation, capillary blood flow and extracellular matrix protein secretion [33, 137, 138]. Moreover, pericytes have an immune-modulatory role in the brain [139]. With reference to the origin of the pericytes, both the mesenchymal and neural crest cells have been proposed to give rise to pericytes in many organs. It has recently been reported that pericytes play important roles in the homeostasis of the brain [140-142]. Pericytes are positioned within the BBB between endothelial cells, astrocytes and neurons [138]. Thus, pericytes can integrate and process signals from their adjacent cells to mediate diverse functional responses that are important for brain function in health and disease, including the regulation of the BBB permeability, angiogenesis, clearance of toxic metabolites, phagocytotic activity, capillary hemodynamic responses and neuroinflammation. ...
A subset of cerebrovascular pericytes originates from mature macrophages in the very early phase of vascular development in CNS
0
2017
Tissue Specific Origin, Development, and Pathological Perspectives of Pericytes
1
2018
... Pericytes are multifunctional cells which ensheath the blood vessel and contribute to the regulation of capillary diameter, vasoconstriction and vasodilation, capillary blood flow and extracellular matrix protein secretion [33, 137, 138]. Moreover, pericytes have an immune-modulatory role in the brain [139]. With reference to the origin of the pericytes, both the mesenchymal and neural crest cells have been proposed to give rise to pericytes in many organs. It has recently been reported that pericytes play important roles in the homeostasis of the brain [140-142]. Pericytes are positioned within the BBB between endothelial cells, astrocytes and neurons [138]. Thus, pericytes can integrate and process signals from their adjacent cells to mediate diverse functional responses that are important for brain function in health and disease, including the regulation of the BBB permeability, angiogenesis, clearance of toxic metabolites, phagocytotic activity, capillary hemodynamic responses and neuroinflammation. ...
Pericyte loss influences Alzheimer-like neurodegeneration in mice
1
2013
... In human post-mortem AD brains and transgenic animal models of AD, pericytes are dysfunctional and they undergo degeneration [143-145]. Also, levels of soluble platelet-derived growth factor receptor β (PDGFR-β), a biomarker for pericyte death, are increased in cerebrospinal fluid (CSF) of subjects with mild dementia and in transgenic AD animals [138, 146, 147]. Thus, there is evidence for pericytes being involved in VaD, although the underlying mechanisms are largely unknown. Nevertheless, a familial form of VaD, CADASIL is associated with dysfunction of pericytes in the brain [148]. ...
Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer's disease
0
2013
Rapid degeneration of cultured human brain pericytes by amyloid beta protein
1
1997
... In human post-mortem AD brains and transgenic animal models of AD, pericytes are dysfunctional and they undergo degeneration [143-145]. Also, levels of soluble platelet-derived growth factor receptor β (PDGFR-β), a biomarker for pericyte death, are increased in cerebrospinal fluid (CSF) of subjects with mild dementia and in transgenic AD animals [138, 146, 147]. Thus, there is evidence for pericytes being involved in VaD, although the underlying mechanisms are largely unknown. Nevertheless, a familial form of VaD, CADASIL is associated with dysfunction of pericytes in the brain [148]. ...
Emerging Biomarkers in Vascular Cognitive Impairment and Dementia: From Pathophysiological Pathways to Clinical Application
2
2019
... In human post-mortem AD brains and transgenic animal models of AD, pericytes are dysfunctional and they undergo degeneration [143-145]. Also, levels of soluble platelet-derived growth factor receptor β (PDGFR-β), a biomarker for pericyte death, are increased in cerebrospinal fluid (CSF) of subjects with mild dementia and in transgenic AD animals [138, 146, 147]. Thus, there is evidence for pericytes being involved in VaD, although the underlying mechanisms are largely unknown. Nevertheless, a familial form of VaD, CADASIL is associated with dysfunction of pericytes in the brain [148]. ...
... Neuroinflammation, like in many other neuro-degenerative diseases, is one of the crucial hallmarks of VaD [146]. It describes a wide range of inflammatory responses within the CNS and is mediated by the production of cytokines, chemokines, and reactive oxygen species. At the cellular level, neuroinflammation involves mainly myeloid cells, i.e. microglia, monocytes, and macrophages, and other peripherally derived immune cells infiltrating the CNS, as well as astrocytes. In response to vascular injuries, microglia are the first cells to respond. Initially classified as M1 pro-inflammatory and anti-regenerative, and M2 anti-inflammatory and pro-regenerative, it is now clear that microglia appear in many different subtypes and phenotypes, with high plasticity and transition between the various types [169-171]. Though microglia have altered activity in VaD even before clinical symptoms and neuropathological hallmarks arise [85, 172], their precise role in VaD is unclear. Nevertheless, under inflammatory conditions, microglia are producers of IL-1b [173], and higher expression of IL-1b has been reported in the brain of hypertensive rats [174]. Also, microglia may contribute to CBF alterations and hypertensive vascular pathology in the brain [175]. Besides microglia, perivascular macrophages might be involved in particular. Monocytes are attracted to the vascular wall by vascular damage, where they differentiate into macrophages, locally secrete matrix proteins, vasculo-protective and pro-angiogenic factors as well as several other growth factors [176], promoting angiogenesis and functional recovery [177]. ...
Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction
1
2019
... In human post-mortem AD brains and transgenic animal models of AD, pericytes are dysfunctional and they undergo degeneration [143-145]. Also, levels of soluble platelet-derived growth factor receptor β (PDGFR-β), a biomarker for pericyte death, are increased in cerebrospinal fluid (CSF) of subjects with mild dementia and in transgenic AD animals [138, 146, 147]. Thus, there is evidence for pericytes being involved in VaD, although the underlying mechanisms are largely unknown. Nevertheless, a familial form of VaD, CADASIL is associated with dysfunction of pericytes in the brain [148]. ...
Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
1
2015
... In human post-mortem AD brains and transgenic animal models of AD, pericytes are dysfunctional and they undergo degeneration [143-145]. Also, levels of soluble platelet-derived growth factor receptor β (PDGFR-β), a biomarker for pericyte death, are increased in cerebrospinal fluid (CSF) of subjects with mild dementia and in transgenic AD animals [138, 146, 147]. Thus, there is evidence for pericytes being involved in VaD, although the underlying mechanisms are largely unknown. Nevertheless, a familial form of VaD, CADASIL is associated with dysfunction of pericytes in the brain [148]. ...
Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies
1
2014
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
TGF-beta1 regulates human brain pericyte inflammatory processes involved in neurovasculature function
2
2016
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
... ]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate
1
1998
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
Interaction between pericytes and endothelial cells leads to formation of tight junction in hyaloid vessels
0
2013
Contextual role for angiopoietins and TGFbeta1 in blood vessel stabilization
1
2007
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
Stem cell quiescence in the hippocampal neurogenic niche is associated with elevated transforming growth factor-beta signaling in an animal model of Huntington disease
5
2010
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
... Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... , 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... ]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
The TGF-beta System As a Potential Pathogenic Player in Disease Modulation of Amyotrophic Lateral Sclerosis
3
2017
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Complement Activation During Ischemia/Reperfusion Injury Induces Pericyte-to-Myofibroblast Transdifferentiation Regulating Peritubular Capillary Lumen Reduction Through pERK Signaling
1
2018
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
Pericytes regulate the blood-brain barrier
1
2010
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
Pericytes are required for blood-brain barrier integrity during embryogenesis
1
2010
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
Apolipoprotein E Inhibits Cerebrovascular Pericyte Mobility through a RhoA Protein-mediated Pathway
1
2015
... Under physiological conditions, TGF-β regulates the proliferation and differentiation of pericytes and their attachment to endothelial cells to facilitate CBF and angiogenesis [33, 137, 149, 150]. Apparently, the interaction between endothelial cells and pericytes leads to the expression of TGF-β by both cell types, with differential responses to the TGF-β activation [151-153]. In fibrosis, hypoxia, brain injury, brain metastasis, neurodegenerative conditions and cerebrovascular pathologies, microglia and astrocytes are activated and produce large amounts of TGF-β1 [70, 74, 154, 155]. Elevated levels of TGF-β in the brain stimulate the proliferation of pericytes and trans-differentiation of pericytes into myofibroblasts [156]. Controversial to that, Rustenhoven et al. have recently described that overexpression of TGF-β suppresses the proliferation of pericytes and reduced the phagocytic ability of pericytes [150]. Moreover, a genetic model of pericyte depletion shows BBB disruption-mediated neuro-degeneration [157, 158]. Besides, a transgenic APOE4 mouse model has signs of BBB abnormality and neurodegeneration associated with dysfunctions of brain pericytes [159]. The aforementioned two genetic models have been shown to exhibit cognitive dysfunctions in association with abnormal TGF-β signaling which might be speculated to result from VaD. Thus, TGF-β mediated pericyte responses in neurocognitive disorders including VaD require detailed scientific investigations. ...
... Many physiological and pathophysiological processes are regulated by extracellular vesicles, which are involved in short and large distance intercellular communications independent of direct cell-to-cell contact [160, 161]. Extracellular vesicles contain high amounts of miRNAs and proteins originated in many different cells including peripheral endothelial cells. At present, exosomes and microvesicles have gained huge attention in neurodegenerative diseases including dementia, for example in AD [162-165]. Of note, in the context of AD and VaD, the intermediate products of amyloid-β, such as lower molecular weight amyloid-β oligomers and protofibrils, are transported in extracellular vesicles spreading from neuron to neuron. Those are neurotoxic and act as a source for further aggregation of amyloid-β in the brain [165]. Interestingly, intracellular amyloid-β is often found to be co-localized with exosomes [166, 167]. Remarkably, mobilization of exosomes is regulated by TGF-β. For example, TGF-β is involved in the transport of MMP2 containing exosomes in malignancy disorders [168]. Therefore, it can be speculated that the increased levels of TGF-β in brains with AD and VaD may play a key role in transport and accumulation of amyloid-β responsible for neurodegeneration. ...
New windows into the brain: Central nervous system-derived extracellular vesicles in blood
1
2019
... Many physiological and pathophysiological processes are regulated by extracellular vesicles, which are involved in short and large distance intercellular communications independent of direct cell-to-cell contact [160, 161]. Extracellular vesicles contain high amounts of miRNAs and proteins originated in many different cells including peripheral endothelial cells. At present, exosomes and microvesicles have gained huge attention in neurodegenerative diseases including dementia, for example in AD [162-165]. Of note, in the context of AD and VaD, the intermediate products of amyloid-β, such as lower molecular weight amyloid-β oligomers and protofibrils, are transported in extracellular vesicles spreading from neuron to neuron. Those are neurotoxic and act as a source for further aggregation of amyloid-β in the brain [165]. Interestingly, intracellular amyloid-β is often found to be co-localized with exosomes [166, 167]. Remarkably, mobilization of exosomes is regulated by TGF-β. For example, TGF-β is involved in the transport of MMP2 containing exosomes in malignancy disorders [168]. Therefore, it can be speculated that the increased levels of TGF-β in brains with AD and VaD may play a key role in transport and accumulation of amyloid-β responsible for neurodegeneration. ...
Extracellular Vesicle-Associated Abeta Mediates Trans-Neuronal Bioenergetic and Ca(2+)-Handling Deficits in Alzheimer's Disease Models
1
2016
... Many physiological and pathophysiological processes are regulated by extracellular vesicles, which are involved in short and large distance intercellular communications independent of direct cell-to-cell contact [160, 161]. Extracellular vesicles contain high amounts of miRNAs and proteins originated in many different cells including peripheral endothelial cells. At present, exosomes and microvesicles have gained huge attention in neurodegenerative diseases including dementia, for example in AD [162-165]. Of note, in the context of AD and VaD, the intermediate products of amyloid-β, such as lower molecular weight amyloid-β oligomers and protofibrils, are transported in extracellular vesicles spreading from neuron to neuron. Those are neurotoxic and act as a source for further aggregation of amyloid-β in the brain [165]. Interestingly, intracellular amyloid-β is often found to be co-localized with exosomes [166, 167]. Remarkably, mobilization of exosomes is regulated by TGF-β. For example, TGF-β is involved in the transport of MMP2 containing exosomes in malignancy disorders [168]. Therefore, it can be speculated that the increased levels of TGF-β in brains with AD and VaD may play a key role in transport and accumulation of amyloid-β responsible for neurodegeneration. ...
Extracellular Vesicle as a Source of Alzheimer's Biomarkers: Opportunities and Challenges
0
2019
The emerging role of exosomes in mental disorders
0
2019
The role of exosomes in the pathogenesis of Alzheimer' disease
2
2017
... Many physiological and pathophysiological processes are regulated by extracellular vesicles, which are involved in short and large distance intercellular communications independent of direct cell-to-cell contact [160, 161]. Extracellular vesicles contain high amounts of miRNAs and proteins originated in many different cells including peripheral endothelial cells. At present, exosomes and microvesicles have gained huge attention in neurodegenerative diseases including dementia, for example in AD [162-165]. Of note, in the context of AD and VaD, the intermediate products of amyloid-β, such as lower molecular weight amyloid-β oligomers and protofibrils, are transported in extracellular vesicles spreading from neuron to neuron. Those are neurotoxic and act as a source for further aggregation of amyloid-β in the brain [165]. Interestingly, intracellular amyloid-β is often found to be co-localized with exosomes [166, 167]. Remarkably, mobilization of exosomes is regulated by TGF-β. For example, TGF-β is involved in the transport of MMP2 containing exosomes in malignancy disorders [168]. Therefore, it can be speculated that the increased levels of TGF-β in brains with AD and VaD may play a key role in transport and accumulation of amyloid-β responsible for neurodegeneration. ...
... ]. Of note, in the context of AD and VaD, the intermediate products of amyloid-β, such as lower molecular weight amyloid-β oligomers and protofibrils, are transported in extracellular vesicles spreading from neuron to neuron. Those are neurotoxic and act as a source for further aggregation of amyloid-β in the brain [165]. Interestingly, intracellular amyloid-β is often found to be co-localized with exosomes [166, 167]. Remarkably, mobilization of exosomes is regulated by TGF-β. For example, TGF-β is involved in the transport of MMP2 containing exosomes in malignancy disorders [168]. Therefore, it can be speculated that the increased levels of TGF-β in brains with AD and VaD may play a key role in transport and accumulation of amyloid-β responsible for neurodegeneration. ...
Exosomes can spread toxic AD pathology
1
2018
... Many physiological and pathophysiological processes are regulated by extracellular vesicles, which are involved in short and large distance intercellular communications independent of direct cell-to-cell contact [160, 161]. Extracellular vesicles contain high amounts of miRNAs and proteins originated in many different cells including peripheral endothelial cells. At present, exosomes and microvesicles have gained huge attention in neurodegenerative diseases including dementia, for example in AD [162-165]. Of note, in the context of AD and VaD, the intermediate products of amyloid-β, such as lower molecular weight amyloid-β oligomers and protofibrils, are transported in extracellular vesicles spreading from neuron to neuron. Those are neurotoxic and act as a source for further aggregation of amyloid-β in the brain [165]. Interestingly, intracellular amyloid-β is often found to be co-localized with exosomes [166, 167]. Remarkably, mobilization of exosomes is regulated by TGF-β. For example, TGF-β is involved in the transport of MMP2 containing exosomes in malignancy disorders [168]. Therefore, it can be speculated that the increased levels of TGF-β in brains with AD and VaD may play a key role in transport and accumulation of amyloid-β responsible for neurodegeneration. ...
Alzheimer's disease pathology propagation by exosomes containing toxic amyloid-beta oligomers
1
2018
... Many physiological and pathophysiological processes are regulated by extracellular vesicles, which are involved in short and large distance intercellular communications independent of direct cell-to-cell contact [160, 161]. Extracellular vesicles contain high amounts of miRNAs and proteins originated in many different cells including peripheral endothelial cells. At present, exosomes and microvesicles have gained huge attention in neurodegenerative diseases including dementia, for example in AD [162-165]. Of note, in the context of AD and VaD, the intermediate products of amyloid-β, such as lower molecular weight amyloid-β oligomers and protofibrils, are transported in extracellular vesicles spreading from neuron to neuron. Those are neurotoxic and act as a source for further aggregation of amyloid-β in the brain [165]. Interestingly, intracellular amyloid-β is often found to be co-localized with exosomes [166, 167]. Remarkably, mobilization of exosomes is regulated by TGF-β. For example, TGF-β is involved in the transport of MMP2 containing exosomes in malignancy disorders [168]. Therefore, it can be speculated that the increased levels of TGF-β in brains with AD and VaD may play a key role in transport and accumulation of amyloid-β responsible for neurodegeneration. ...
TGF-beta-mediated exosomal lnc-MMP2-2 regulates migration and invasion of lung cancer cells to the vasculature by promoting MMP2 expression
1
2018
... Many physiological and pathophysiological processes are regulated by extracellular vesicles, which are involved in short and large distance intercellular communications independent of direct cell-to-cell contact [160, 161]. Extracellular vesicles contain high amounts of miRNAs and proteins originated in many different cells including peripheral endothelial cells. At present, exosomes and microvesicles have gained huge attention in neurodegenerative diseases including dementia, for example in AD [162-165]. Of note, in the context of AD and VaD, the intermediate products of amyloid-β, such as lower molecular weight amyloid-β oligomers and protofibrils, are transported in extracellular vesicles spreading from neuron to neuron. Those are neurotoxic and act as a source for further aggregation of amyloid-β in the brain [165]. Interestingly, intracellular amyloid-β is often found to be co-localized with exosomes [166, 167]. Remarkably, mobilization of exosomes is regulated by TGF-β. For example, TGF-β is involved in the transport of MMP2 containing exosomes in malignancy disorders [168]. Therefore, it can be speculated that the increased levels of TGF-β in brains with AD and VaD may play a key role in transport and accumulation of amyloid-β responsible for neurodegeneration. ...
Neuroinflammation and M2 microglia: the good, the bad, and the inflamed
1
2014
... Neuroinflammation, like in many other neuro-degenerative diseases, is one of the crucial hallmarks of VaD [146]. It describes a wide range of inflammatory responses within the CNS and is mediated by the production of cytokines, chemokines, and reactive oxygen species. At the cellular level, neuroinflammation involves mainly myeloid cells, i.e. microglia, monocytes, and macrophages, and other peripherally derived immune cells infiltrating the CNS, as well as astrocytes. In response to vascular injuries, microglia are the first cells to respond. Initially classified as M1 pro-inflammatory and anti-regenerative, and M2 anti-inflammatory and pro-regenerative, it is now clear that microglia appear in many different subtypes and phenotypes, with high plasticity and transition between the various types [169-171]. Though microglia have altered activity in VaD even before clinical symptoms and neuropathological hallmarks arise [85, 172], their precise role in VaD is unclear. Nevertheless, under inflammatory conditions, microglia are producers of IL-1b [173], and higher expression of IL-1b has been reported in the brain of hypertensive rats [174]. Also, microglia may contribute to CBF alterations and hypertensive vascular pathology in the brain [175]. Besides microglia, perivascular macrophages might be involved in particular. Monocytes are attracted to the vascular wall by vascular damage, where they differentiate into macrophages, locally secrete matrix proteins, vasculo-protective and pro-angiogenic factors as well as several other growth factors [176], promoting angiogenesis and functional recovery [177]. ...
Dynamic changes to lipid mediators support transitions among macrophage subtypes during muscle regeneration
0
2019
Microglia emerge as central players in brain disease
1
2017
... Neuroinflammation, like in many other neuro-degenerative diseases, is one of the crucial hallmarks of VaD [146]. It describes a wide range of inflammatory responses within the CNS and is mediated by the production of cytokines, chemokines, and reactive oxygen species. At the cellular level, neuroinflammation involves mainly myeloid cells, i.e. microglia, monocytes, and macrophages, and other peripherally derived immune cells infiltrating the CNS, as well as astrocytes. In response to vascular injuries, microglia are the first cells to respond. Initially classified as M1 pro-inflammatory and anti-regenerative, and M2 anti-inflammatory and pro-regenerative, it is now clear that microglia appear in many different subtypes and phenotypes, with high plasticity and transition between the various types [169-171]. Though microglia have altered activity in VaD even before clinical symptoms and neuropathological hallmarks arise [85, 172], their precise role in VaD is unclear. Nevertheless, under inflammatory conditions, microglia are producers of IL-1b [173], and higher expression of IL-1b has been reported in the brain of hypertensive rats [174]. Also, microglia may contribute to CBF alterations and hypertensive vascular pathology in the brain [175]. Besides microglia, perivascular macrophages might be involved in particular. Monocytes are attracted to the vascular wall by vascular damage, where they differentiate into macrophages, locally secrete matrix proteins, vasculo-protective and pro-angiogenic factors as well as several other growth factors [176], promoting angiogenesis and functional recovery [177]. ...
Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease
1
2017
... Neuroinflammation, like in many other neuro-degenerative diseases, is one of the crucial hallmarks of VaD [146]. It describes a wide range of inflammatory responses within the CNS and is mediated by the production of cytokines, chemokines, and reactive oxygen species. At the cellular level, neuroinflammation involves mainly myeloid cells, i.e. microglia, monocytes, and macrophages, and other peripherally derived immune cells infiltrating the CNS, as well as astrocytes. In response to vascular injuries, microglia are the first cells to respond. Initially classified as M1 pro-inflammatory and anti-regenerative, and M2 anti-inflammatory and pro-regenerative, it is now clear that microglia appear in many different subtypes and phenotypes, with high plasticity and transition between the various types [169-171]. Though microglia have altered activity in VaD even before clinical symptoms and neuropathological hallmarks arise [85, 172], their precise role in VaD is unclear. Nevertheless, under inflammatory conditions, microglia are producers of IL-1b [173], and higher expression of IL-1b has been reported in the brain of hypertensive rats [174]. Also, microglia may contribute to CBF alterations and hypertensive vascular pathology in the brain [175]. Besides microglia, perivascular macrophages might be involved in particular. Monocytes are attracted to the vascular wall by vascular damage, where they differentiate into macrophages, locally secrete matrix proteins, vasculo-protective and pro-angiogenic factors as well as several other growth factors [176], promoting angiogenesis and functional recovery [177]. ...
Requirement for interleukin-1 to drive brain inflammation reveals tissue-specific mechanisms of innate immunity
1
2015
... Neuroinflammation, like in many other neuro-degenerative diseases, is one of the crucial hallmarks of VaD [146]. It describes a wide range of inflammatory responses within the CNS and is mediated by the production of cytokines, chemokines, and reactive oxygen species. At the cellular level, neuroinflammation involves mainly myeloid cells, i.e. microglia, monocytes, and macrophages, and other peripherally derived immune cells infiltrating the CNS, as well as astrocytes. In response to vascular injuries, microglia are the first cells to respond. Initially classified as M1 pro-inflammatory and anti-regenerative, and M2 anti-inflammatory and pro-regenerative, it is now clear that microglia appear in many different subtypes and phenotypes, with high plasticity and transition between the various types [169-171]. Though microglia have altered activity in VaD even before clinical symptoms and neuropathological hallmarks arise [85, 172], their precise role in VaD is unclear. Nevertheless, under inflammatory conditions, microglia are producers of IL-1b [173], and higher expression of IL-1b has been reported in the brain of hypertensive rats [174]. Also, microglia may contribute to CBF alterations and hypertensive vascular pathology in the brain [175]. Besides microglia, perivascular macrophages might be involved in particular. Monocytes are attracted to the vascular wall by vascular damage, where they differentiate into macrophages, locally secrete matrix proteins, vasculo-protective and pro-angiogenic factors as well as several other growth factors [176], promoting angiogenesis and functional recovery [177]. ...
Neuroinflammatory Markers in Spontaneously Hypertensive Rat Brain: An Immunohistochemical Study
1
2016
... Neuroinflammation, like in many other neuro-degenerative diseases, is one of the crucial hallmarks of VaD [146]. It describes a wide range of inflammatory responses within the CNS and is mediated by the production of cytokines, chemokines, and reactive oxygen species. At the cellular level, neuroinflammation involves mainly myeloid cells, i.e. microglia, monocytes, and macrophages, and other peripherally derived immune cells infiltrating the CNS, as well as astrocytes. In response to vascular injuries, microglia are the first cells to respond. Initially classified as M1 pro-inflammatory and anti-regenerative, and M2 anti-inflammatory and pro-regenerative, it is now clear that microglia appear in many different subtypes and phenotypes, with high plasticity and transition between the various types [169-171]. Though microglia have altered activity in VaD even before clinical symptoms and neuropathological hallmarks arise [85, 172], their precise role in VaD is unclear. Nevertheless, under inflammatory conditions, microglia are producers of IL-1b [173], and higher expression of IL-1b has been reported in the brain of hypertensive rats [174]. Also, microglia may contribute to CBF alterations and hypertensive vascular pathology in the brain [175]. Besides microglia, perivascular macrophages might be involved in particular. Monocytes are attracted to the vascular wall by vascular damage, where they differentiate into macrophages, locally secrete matrix proteins, vasculo-protective and pro-angiogenic factors as well as several other growth factors [176], promoting angiogenesis and functional recovery [177]. ...
The importance of comorbidities in ischemic stroke: Impact of hypertension on the cerebral circulation
1
2018
... Neuroinflammation, like in many other neuro-degenerative diseases, is one of the crucial hallmarks of VaD [146]. It describes a wide range of inflammatory responses within the CNS and is mediated by the production of cytokines, chemokines, and reactive oxygen species. At the cellular level, neuroinflammation involves mainly myeloid cells, i.e. microglia, monocytes, and macrophages, and other peripherally derived immune cells infiltrating the CNS, as well as astrocytes. In response to vascular injuries, microglia are the first cells to respond. Initially classified as M1 pro-inflammatory and anti-regenerative, and M2 anti-inflammatory and pro-regenerative, it is now clear that microglia appear in many different subtypes and phenotypes, with high plasticity and transition between the various types [169-171]. Though microglia have altered activity in VaD even before clinical symptoms and neuropathological hallmarks arise [85, 172], their precise role in VaD is unclear. Nevertheless, under inflammatory conditions, microglia are producers of IL-1b [173], and higher expression of IL-1b has been reported in the brain of hypertensive rats [174]. Also, microglia may contribute to CBF alterations and hypertensive vascular pathology in the brain [175]. Besides microglia, perivascular macrophages might be involved in particular. Monocytes are attracted to the vascular wall by vascular damage, where they differentiate into macrophages, locally secrete matrix proteins, vasculo-protective and pro-angiogenic factors as well as several other growth factors [176], promoting angiogenesis and functional recovery [177]. ...
Macrophages prevent hemorrhagic infarct transformation in murine stroke models
1
2012
... Neuroinflammation, like in many other neuro-degenerative diseases, is one of the crucial hallmarks of VaD [146]. It describes a wide range of inflammatory responses within the CNS and is mediated by the production of cytokines, chemokines, and reactive oxygen species. At the cellular level, neuroinflammation involves mainly myeloid cells, i.e. microglia, monocytes, and macrophages, and other peripherally derived immune cells infiltrating the CNS, as well as astrocytes. In response to vascular injuries, microglia are the first cells to respond. Initially classified as M1 pro-inflammatory and anti-regenerative, and M2 anti-inflammatory and pro-regenerative, it is now clear that microglia appear in many different subtypes and phenotypes, with high plasticity and transition between the various types [169-171]. Though microglia have altered activity in VaD even before clinical symptoms and neuropathological hallmarks arise [85, 172], their precise role in VaD is unclear. Nevertheless, under inflammatory conditions, microglia are producers of IL-1b [173], and higher expression of IL-1b has been reported in the brain of hypertensive rats [174]. Also, microglia may contribute to CBF alterations and hypertensive vascular pathology in the brain [175]. Besides microglia, perivascular macrophages might be involved in particular. Monocytes are attracted to the vascular wall by vascular damage, where they differentiate into macrophages, locally secrete matrix proteins, vasculo-protective and pro-angiogenic factors as well as several other growth factors [176], promoting angiogenesis and functional recovery [177]. ...
Monocyte-Derived Macrophages Contribute to Spontaneous Long-Term Functional Recovery after Stroke in Mice
1
2016
... Neuroinflammation, like in many other neuro-degenerative diseases, is one of the crucial hallmarks of VaD [146]. It describes a wide range of inflammatory responses within the CNS and is mediated by the production of cytokines, chemokines, and reactive oxygen species. At the cellular level, neuroinflammation involves mainly myeloid cells, i.e. microglia, monocytes, and macrophages, and other peripherally derived immune cells infiltrating the CNS, as well as astrocytes. In response to vascular injuries, microglia are the first cells to respond. Initially classified as M1 pro-inflammatory and anti-regenerative, and M2 anti-inflammatory and pro-regenerative, it is now clear that microglia appear in many different subtypes and phenotypes, with high plasticity and transition between the various types [169-171]. Though microglia have altered activity in VaD even before clinical symptoms and neuropathological hallmarks arise [85, 172], their precise role in VaD is unclear. Nevertheless, under inflammatory conditions, microglia are producers of IL-1b [173], and higher expression of IL-1b has been reported in the brain of hypertensive rats [174]. Also, microglia may contribute to CBF alterations and hypertensive vascular pathology in the brain [175]. Besides microglia, perivascular macrophages might be involved in particular. Monocytes are attracted to the vascular wall by vascular damage, where they differentiate into macrophages, locally secrete matrix proteins, vasculo-protective and pro-angiogenic factors as well as several other growth factors [176], promoting angiogenesis and functional recovery [177]. ...
Silencing of TGFbeta signalling in microglia results in impaired homeostasis
2
2018
... Neuroinflammation, in particular, microglia activity is strongly regulated by TGF-β. It seems that TGF-β signaling maintains microglia homeostasis. For example, conditional loss of TGF-β signaling in microglia resulted in upregulation of activation and priming markers indicative of a pro-inflammatory stage [178]. Also, TGF-β1 modulates the microglial phenotype and promotes recovery after intracerebral hemorrhage [179]. In the context of VaD, TGF-β regulates microglial activity and induces resistance to hypertension. For example, removal of TGF-β or blocking its signaling before hypertension induction accelerated hypertension progression, and supplementation of TGF-β1 substantially suppressed neuroinflammation and generated immunosuppressive microglia [180]. In summary, TGF-β has, in the context of neuroinflammation and microglia, anti-inflammatory and homeostatic functions (Fig. 4). ...
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
TGF-beta1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage
1
2017
... Neuroinflammation, in particular, microglia activity is strongly regulated by TGF-β. It seems that TGF-β signaling maintains microglia homeostasis. For example, conditional loss of TGF-β signaling in microglia resulted in upregulation of activation and priming markers indicative of a pro-inflammatory stage [178]. Also, TGF-β1 modulates the microglial phenotype and promotes recovery after intracerebral hemorrhage [179]. In the context of VaD, TGF-β regulates microglial activity and induces resistance to hypertension. For example, removal of TGF-β or blocking its signaling before hypertension induction accelerated hypertension progression, and supplementation of TGF-β1 substantially suppressed neuroinflammation and generated immunosuppressive microglia [180]. In summary, TGF-β has, in the context of neuroinflammation and microglia, anti-inflammatory and homeostatic functions (Fig. 4). ...
Brain Transforming Growth Factor-beta Resists Hypertension Via Regulating Microglial Activation
1
2017
... Neuroinflammation, in particular, microglia activity is strongly regulated by TGF-β. It seems that TGF-β signaling maintains microglia homeostasis. For example, conditional loss of TGF-β signaling in microglia resulted in upregulation of activation and priming markers indicative of a pro-inflammatory stage [178]. Also, TGF-β1 modulates the microglial phenotype and promotes recovery after intracerebral hemorrhage [179]. In the context of VaD, TGF-β regulates microglial activity and induces resistance to hypertension. For example, removal of TGF-β or blocking its signaling before hypertension induction accelerated hypertension progression, and supplementation of TGF-β1 substantially suppressed neuroinflammation and generated immunosuppressive microglia [180]. In summary, TGF-β has, in the context of neuroinflammation and microglia, anti-inflammatory and homeostatic functions (Fig. 4). ...
The neglected co-star in the dementia drama: the putative roles of astrocytes in the pathogeneses of major neurocognitive disorders
1
2014
... Besides monocytes and macrophages, astrocytes are involved in the pathology of VaD. For example, astrogliosis is an acute (7 days) and chronic (3 months) response to artery occlusion in rats [181-183]. Here, in contrast to the effects on microglia, TGF-β seems to have detrimental effects. For example, TGF-β induces astrogliosis and is the main driver of gliotic scarring [74]. In the context of VaD, TGF-β overexpression induces a thickened vascular wall due to accumulation of extracellular matrix proteins [184]. In summary, TGF-β plays a crucial role in neuroinflammation, in particular, in the context of microglial and astroglial cells. ...
An emerging role of astrocytes in vascular contributions to cognitive impairment and dementia
0
2018
Prevention of Hippocampal Neuronal Damage and Cognitive Function Deficits in Vascular Dementia by Dextromethorphan
1
2016
... Besides monocytes and macrophages, astrocytes are involved in the pathology of VaD. For example, astrogliosis is an acute (7 days) and chronic (3 months) response to artery occlusion in rats [181-183]. Here, in contrast to the effects on microglia, TGF-β seems to have detrimental effects. For example, TGF-β induces astrogliosis and is the main driver of gliotic scarring [74]. In the context of VaD, TGF-β overexpression induces a thickened vascular wall due to accumulation of extracellular matrix proteins [184]. In summary, TGF-β plays a crucial role in neuroinflammation, in particular, in the context of microglial and astroglial cells. ...
Intact memory in TGF-beta1 transgenic mice featuring chronic cerebrovascular deficit: recovery with pioglitazone
1
2011
... Besides monocytes and macrophages, astrocytes are involved in the pathology of VaD. For example, astrogliosis is an acute (7 days) and chronic (3 months) response to artery occlusion in rats [181-183]. Here, in contrast to the effects on microglia, TGF-β seems to have detrimental effects. For example, TGF-β induces astrogliosis and is the main driver of gliotic scarring [74]. In the context of VaD, TGF-β overexpression induces a thickened vascular wall due to accumulation of extracellular matrix proteins [184]. In summary, TGF-β plays a crucial role in neuroinflammation, in particular, in the context of microglial and astroglial cells. ...
Staging and natural history of cerebrovascular pathology in dementia
1
2012
... White matter lesions are defined by white matter hyperintensities in magnetic resonance imaging and are a hallmark of VaD. At the cellular level, white matter lesions are characterized by myelin and oligodendrocyte loss, by axonal damages associated with axonal thinning and the accumulation of varicosities, and by loosening of the axon-oligodendrocyte adhesion [185-188]. Therefore, myelin protection and remyelination are clear targets in VaD. This is, in particular, the case in the aged brain, where remyelination is insufficient [189, 190]. ...
Progression of white matter hyperintensities and incidence of new lacunes over a 3-year period: the Leukoaraiosis and Disability study
0
2008
Molecular disorganization of axons adjacent to human lacunar infarcts
0
2015
Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment
1
2016
... White matter lesions are defined by white matter hyperintensities in magnetic resonance imaging and are a hallmark of VaD. At the cellular level, white matter lesions are characterized by myelin and oligodendrocyte loss, by axonal damages associated with axonal thinning and the accumulation of varicosities, and by loosening of the axon-oligodendrocyte adhesion [185-188]. Therefore, myelin protection and remyelination are clear targets in VaD. This is, in particular, the case in the aged brain, where remyelination is insufficient [189, 190]. ...
Aging restricts the ability of mesenchymal stem cells to promote the generation of oligodendrocytes during remyelination
1
2019
... White matter lesions are defined by white matter hyperintensities in magnetic resonance imaging and are a hallmark of VaD. At the cellular level, white matter lesions are characterized by myelin and oligodendrocyte loss, by axonal damages associated with axonal thinning and the accumulation of varicosities, and by loosening of the axon-oligodendrocyte adhesion [185-188]. Therefore, myelin protection and remyelination are clear targets in VaD. This is, in particular, the case in the aged brain, where remyelination is insufficient [189, 190]. ...
Rejuvenation of regeneration in the aging central nervous system
1
2012
... White matter lesions are defined by white matter hyperintensities in magnetic resonance imaging and are a hallmark of VaD. At the cellular level, white matter lesions are characterized by myelin and oligodendrocyte loss, by axonal damages associated with axonal thinning and the accumulation of varicosities, and by loosening of the axon-oligodendrocyte adhesion [185-188]. Therefore, myelin protection and remyelination are clear targets in VaD. This is, in particular, the case in the aged brain, where remyelination is insufficient [189, 190]. ...
TGF-beta1 is associated with the progression of intracranial deep white matter lesions: a pilot study with 5 years of magnetic resonance imaging follow-up
1
2014
... The role of TGF-β in de- and remyelination is still controversially discussed. For example, TGF-β1 is associated with the progression of intracranial deep white matter lesions in humans [191]. In contrast, TGF-β1, which is present in higher levels in the systemic environment, promotes oligodendrocyte maturation, while lower levels in circulating TGF-β prevent remyelination in the spinal cord after toxin-induced demyelination [192]. Also, TGF-β promoted subcortical white matter remyelination, while TGF-β receptor deletion in oligodendroglial progenitors prevents their development into myelinating oligodendrocytes [193]. ...
Circulating transforming growth factor-beta1 facilitates remyelination in the adult central nervous system
1
2019
... The role of TGF-β in de- and remyelination is still controversially discussed. For example, TGF-β1 is associated with the progression of intracranial deep white matter lesions in humans [191]. In contrast, TGF-β1, which is present in higher levels in the systemic environment, promotes oligodendrocyte maturation, while lower levels in circulating TGF-β prevent remyelination in the spinal cord after toxin-induced demyelination [192]. Also, TGF-β promoted subcortical white matter remyelination, while TGF-β receptor deletion in oligodendroglial progenitors prevents their development into myelinating oligodendrocytes [193]. ...
TGFbeta signaling regulates the timing of CNS myelination by modulating oligodendrocyte progenitor cell cycle exit through SMAD3/4/FoxO1/Sp1
1
2014
... The role of TGF-β in de- and remyelination is still controversially discussed. For example, TGF-β1 is associated with the progression of intracranial deep white matter lesions in humans [191]. In contrast, TGF-β1, which is present in higher levels in the systemic environment, promotes oligodendrocyte maturation, while lower levels in circulating TGF-β prevent remyelination in the spinal cord after toxin-induced demyelination [192]. Also, TGF-β promoted subcortical white matter remyelination, while TGF-β receptor deletion in oligodendroglial progenitors prevents their development into myelinating oligodendrocytes [193]. ...
Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity
1
1990
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Glial Contribution to Excitatory and Inhibitory Synapse Loss in Neurodegeneration
0
2019
Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment
1
1991
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Synaptic markers of cognitive decline in neurodegenerative diseases: a proteomic approach
1
2018
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Synaptophysin I controls the targeting of VAMP2/synaptobrevin II to synaptic vesicles
0
2003
Synaptic Plasticity, Dementia and Alzheimer Disease
2
2017
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
... ]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease
1
1997
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
PSD-95 is required to sustain the molecular organization of the postsynaptic density
1
2011
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Postsynaptic density protein-95 regulates NMDA channel gating and surface expression
1
2004
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Synaptic protein levels altered in vascular dementia
4
2015
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
... , 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
... , 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
... ]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer's disease
1
2010
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
SNAP-25, a Known Presynaptic Protein with Emerging Postsynaptic Functions
1
2016
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development
1
1995
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Neurodegeneration and Alzheimer's disease (AD). What Can Proteomics Tell Us About the Alzheimer's Brain?
1
2016
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Down-regulation of drebrin A expression suppresses synaptic targeting of NMDA receptors in developing hippocampal neurones
1
2006
... Synaptic loss and neurodegeneration are the foremost neuropathological hallmarks that reflect various forms of dementia [194-196]. With reference to synaptic function, a wide range of proteins involved in vesicular trafficking, synapse structure and signal transduction are known. Synaptophysin, a presynaptic vesicle-specific protein is considered as a marker of synaptic integrity [197-199]. Protein expression studies on synaptic membrane preparations from various brain regions of subjects with AD and VaD revealed a reduction of synaptophysin levels in the hippocampus [200]. Postsynaptic density protein 95 (PSD-95) is a membrane-associated guanylate cyclase family of proteins located in dendritic spines and is required for synaptic plasticity [201, 202]. The expression of PSD-95 is reduced significantly in AD, mild cognitive impairment and VaD [203, 204]. Synaptosomal-associated protein 25 (SNAP-25) is part of the SNARE complex involved in synaptic vesicle membrane docking and fusion, and is present in the presynaptic terminal participating in exocytosis and neurotransmitter release [205, 206]. A significant reduction in the expression of SNAP-25 is reported in AD brains, in frontotemporal dementia and VaD [199, 203, 207]. Drebrin (developmentally regulated brain protein), an F-actin binding protein involved in dendritic spine morphogenesis, is also drastically reduced in AD and mild cognitive impairment but increased in VaD [17, 203, 208]. The rationality of the increased level of drebrin in VaD is not clear but it might be a compensatory response to the ischemia related pathomechanisms caused by VaD [203]. The dysregulation in the expression of presynaptic and synaptic proteins that are involved in synaptic function are correlated with a significant loss of synapses in the brains of subjects with different forms of dementia. ...
Impaired glutamate recycling and GluN2B-mediated neuronal calcium overload in mice lacking TGF-beta1 in the CNS
1
2013
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
A key role for TGF-beta1 in hippocampal synaptic plasticity and memory
1
2015
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
Transforming growth factor beta1 alters synapsin distribution and modulates synaptic depression in Aplysia
1
2002
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain
1
2003
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
The neuroprotective functions of transforming growth factor beta proteins
1
2012
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
The role of TGF-beta superfamily signaling in neurological disorders
0
2018
Transforming growth factor beta promotes neuronal cell fate of mouse cortical and hippocampal progenitors in vitro and in vivo: identification of Nedd9 as an essential signaling component
1
2010
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
Transforming growth factor-beta 1 potentiates amyloid-beta generation in astrocytes and in transgenic mice
1
2003
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
Role of the Transforming-Growth-Factor-beta1 Gene in Late-Onset Alzheimer's Disease: Implications for the Treatment
1
2013
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
Comparing Effects of Transforming Growth Factor beta1 on Microglia From Rat and Mouse: Transcriptional Profiles and Potassium Channels
0
2018
Role of TGFbeta signaling in the pathogenesis of Alzheimer's disease
1
2015
... Several studies have indicated that the induction of TGF-β during pathogenesis exerts a defensive role in preventing neurodegeneration [219]. Reduced expression of TGF-β and deficient Smad signaling in neurons induces neurodegeneration in the terminal stage of the disease [61]. The reduced neurogenesis seen in the brains of some models of AD could provide a possible explanation for the reduction in TGF-β mediated neurodegeneration in cognitive centers of the pathological brain. Recently, reactive neuroblasts in the diseased brain have been proposed to acquire properties of non-neuronal cells such as microglia, and thus these cells might represent a potential source of TGF-β in pathological brains [223, 253]. The robust activation, proliferation and circulation of microglia, reactive astrocytes and neuroblasts found in early pathogenesis tend to be diminished in the late phases of the diseases [227, 254]. Thus, it can be hypothesized that the overall loss of neuroblasts, non-neuronal and neuronal cells in the brain in the late phase of the disease might lead to the reduced level of TGF-β, thereby fostering neurodegeneration and memory loss. ...
Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia
1
2018
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
Synaptic dysfunction in Alzheimer's disease
1
2012
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
Neurodegenerative disease: Proteome points to synaptic dysfunction in dementia
1
2018
... A role of TGF-β in synaptic protein expression is not known, but TGF-β certainly modulates spines and spine densities. For example, the brain-specific knock down of TGF-β1 in mice showed a reduction in the dendritic spine density and a defect in neurotransmission in the hippocampus [209]. Treatment of the sea-snail Aplysia with TGF-β1 promotes sensorimotor synapses and reduces synaptic dysfunctions through the activation of MAPK and BDNF-mediated CREB phosphorylation [210, 211]. Also, deficiencies in the expression of TGF-β, deletion of TGF-β receptors and pharmacological inhibition of Smad2/3 phosphorylation induces neurodegeneration and synaptic dysfunctions [54, 178, 212-215]. Astroglial-specific production of TGF-β in aged transgenic mice expressing the amyloid beta precursor protein (APP) minimizes dystrophic neurites and rescues neurodegeneration [51, 216]. However, prolonged activation of microglia and thereby induced presence of an increased amount of TGF-β in the brain is linked to the synaptic loss in AD and VaD [155, 217-220]. Also of relevance, the clinical signature of various forms of dementia is most likely linked to the synaptic dysfunctions rather than neurodegeneration [221, 222]. ...
Neuroplasticity, limbic neuroblastosis and neuro-regenerative disorders
3
2018
... Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... Several studies have indicated that the induction of TGF-β during pathogenesis exerts a defensive role in preventing neurodegeneration [219]. Reduced expression of TGF-β and deficient Smad signaling in neurons induces neurodegeneration in the terminal stage of the disease [61]. The reduced neurogenesis seen in the brains of some models of AD could provide a possible explanation for the reduction in TGF-β mediated neurodegeneration in cognitive centers of the pathological brain. Recently, reactive neuroblasts in the diseased brain have been proposed to acquire properties of non-neuronal cells such as microglia, and thus these cells might represent a potential source of TGF-β in pathological brains [223, 253]. The robust activation, proliferation and circulation of microglia, reactive astrocytes and neuroblasts found in early pathogenesis tend to be diminished in the late phases of the diseases [227, 254]. Thus, it can be hypothesized that the overall loss of neuroblasts, non-neuronal and neuronal cells in the brain in the late phase of the disease might lead to the reduced level of TGF-β, thereby fostering neurodegeneration and memory loss. ...
A Milieu Molecule for TGF-beta Required for Microglia Function in the Nervous System
1
2018
... Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
Transforming growth factor-beta1 is a negative modulator of adult neurogenesis
2
2006
... Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... , 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
Vascular-derived TGF-beta increases in the stem cell niche and perturbs neurogenesis during aging and following irradiation in the adult mouse brain
1
2013
... Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
Reactive Neuroblastosis in Huntington's Disease: A Putative Therapeutic Target for Striatal Regeneration in the Adult Brain
5
2018
... Defects in hippocampal neurogenesis are proposed to contribute to cognitive deficits in dementia [223]. Recently, we provided experimental evidence for the widespread expression of the TGF-β signaling components including Smad in the intact adult brain [38]. Immunoblotting and immunocytochemistry analyses revealed that considerable expression of TGF-βR2 is evident in the olfactory bulb cortex, striatum and cerebellum of the adult rat brain. Expression of the TGF-βR2 is relatively low in the neurogenic niches such as the hippocampus and the SVZ. However, we have noticed high expression levels of TGF-βR1 as well as the phosphorylated form of Smad2 (pSmad2) throughout the adult brain including the neurogenic regions [38]. Further, extensive investigation of the pSmad2 expression in the hippocampal neurogenic niche using co-immunolabeling analysis revealed that proliferating subpopulations of Sox2 positive neural stem cells, DCX-positive neuroblasts and GFAP-positive astrocytes failed to show immunoreactivity for pSmad2, suggesting that they were not under TGF-β stimulation. However, mitotically inactive NSCs, neuroblasts as well as mature neurons exhibit a high-level nuclear expression of pSmad2 in the adult hippocampus [38, 154]. Strikingly, glial cells such as GFAP-positive astrocytes and IBA-1 positive microglia are characterized by the expression of TGF-β [76, 224]. However, these immunological cells of the brain appear to be devoid of pSmad2 [38, 58]. Thus, it suggests that glial cell-specific TGF-β mediates paracrine signaling in the NSC niche rather than autocrine signaling in the adult brain. Further, we observed the direct effect of TGF-β on the regulation of neurogenesis in the brain through intraventricular infusion or a transgenic system expressing TGF-β in the brain under the doxycycline-controlled Ca-Calmodulin kinase promoter [38, 58, 225]. Elevation of TGF-β reduced proliferation of NSCs but promoted the survival of neuroblasts [38]. Besides, in an in vitro study, incubation of neurosphere cultures with TGF-β pushed the cells into the G0 phase of the cell cycle [38, 225]. Subsequently, the above-mentioned facts were confirmed in the brains of transgenic animals that overexpress TGF-β1 under the control of the GFAP promoter in astrocytes [77]. Taken together, it has been hypothesized that TGF-β mediated pSmad2 signaling is responsible for the maintenance of NSCs by inducing cellular quiescence as well as the determination of neural lineage of NSCs and neuroprotection. Moreover, as the expression of TGF-β increases in the aging brain, it might contribute to a decline in neurogenesis [54]. There are some studies indicate that overexpression of TGF-β can induce cell death in NSCs in the hippocampus of the adult brain [226]. Notably, aging brains and many neurocognitive disorders have been characterized by pathologically elevated levels of TGF-β and defects in regenerative plasticity [227]. Hence, it has been conceptualized that impaired neurogenesis in aging and dementia can be attributed to the pathomechanism of aberrant TGF β pathway. ...
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... , 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... ]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... Several studies have indicated that the induction of TGF-β during pathogenesis exerts a defensive role in preventing neurodegeneration [219]. Reduced expression of TGF-β and deficient Smad signaling in neurons induces neurodegeneration in the terminal stage of the disease [61]. The reduced neurogenesis seen in the brains of some models of AD could provide a possible explanation for the reduction in TGF-β mediated neurodegeneration in cognitive centers of the pathological brain. Recently, reactive neuroblasts in the diseased brain have been proposed to acquire properties of non-neuronal cells such as microglia, and thus these cells might represent a potential source of TGF-β in pathological brains [223, 253]. The robust activation, proliferation and circulation of microglia, reactive astrocytes and neuroblasts found in early pathogenesis tend to be diminished in the late phases of the diseases [227, 254]. Thus, it can be hypothesized that the overall loss of neuroblasts, non-neuronal and neuronal cells in the brain in the late phase of the disease might lead to the reduced level of TGF-β, thereby fostering neurodegeneration and memory loss. ...
Animal Models of Vascular Cognitive Impairment and Dementia (VCID)
1
2016
... In an animal model of VaD, a 2-vessel occlusion (2VO) in experimental rats mimicking chronic cerebral hypoperfusion (CCH) [228, 229], hippocampal neurogenesis is more severely reduced in association with cognitive impairments [230]. Physical exercise rescues the deficits in hippocampal neurogenesis and cognitive functions through the BDNF-CREB pathway in 2VO models [231]. In another study reported by Kwon et al., the inhibition of acetylcholinesterase promotes hippocampal neurogenesis and cognitive function in VaD [232]. Moreover, transplantation of MSCs provided neuroprotection in the hippocampus of 2VO models [233]. Reports on the effects of VaD induced TGF-β levels on adult neurogenesis are limited. As many animal models of AD display a vascular pathology, the regulation of adult neurogenesis by TGF-β can be expected to be similar in VaD as it is described in AD [234, 235]. In corroboration with existing findings on AD and reports generated from experimental models of VaD, it can be presumed that abnormal levels of TGF-β in association with impaired neurogenesis might provide a potential mechanism for vascular abnormalities mediated defects in regenerative plasticity (Fig. 4). ...
Progressive cognitive impairment following chronic cerebral hypoperfusion induced by permanent occlusion of bilateral carotid arteries in rats
1
1994
... In an animal model of VaD, a 2-vessel occlusion (2VO) in experimental rats mimicking chronic cerebral hypoperfusion (CCH) [228, 229], hippocampal neurogenesis is more severely reduced in association with cognitive impairments [230]. Physical exercise rescues the deficits in hippocampal neurogenesis and cognitive functions through the BDNF-CREB pathway in 2VO models [231]. In another study reported by Kwon et al., the inhibition of acetylcholinesterase promotes hippocampal neurogenesis and cognitive function in VaD [232]. Moreover, transplantation of MSCs provided neuroprotection in the hippocampus of 2VO models [233]. Reports on the effects of VaD induced TGF-β levels on adult neurogenesis are limited. As many animal models of AD display a vascular pathology, the regulation of adult neurogenesis by TGF-β can be expected to be similar in VaD as it is described in AD [234, 235]. In corroboration with existing findings on AD and reports generated from experimental models of VaD, it can be presumed that abnormal levels of TGF-β in association with impaired neurogenesis might provide a potential mechanism for vascular abnormalities mediated defects in regenerative plasticity (Fig. 4). ...
Age-dependent impairment of hippocampal neurogenesis in chronic cerebral hypoperfusion
1
2008
... In an animal model of VaD, a 2-vessel occlusion (2VO) in experimental rats mimicking chronic cerebral hypoperfusion (CCH) [228, 229], hippocampal neurogenesis is more severely reduced in association with cognitive impairments [230]. Physical exercise rescues the deficits in hippocampal neurogenesis and cognitive functions through the BDNF-CREB pathway in 2VO models [231]. In another study reported by Kwon et al., the inhibition of acetylcholinesterase promotes hippocampal neurogenesis and cognitive function in VaD [232]. Moreover, transplantation of MSCs provided neuroprotection in the hippocampus of 2VO models [233]. Reports on the effects of VaD induced TGF-β levels on adult neurogenesis are limited. As many animal models of AD display a vascular pathology, the regulation of adult neurogenesis by TGF-β can be expected to be similar in VaD as it is described in AD [234, 235]. In corroboration with existing findings on AD and reports generated from experimental models of VaD, it can be presumed that abnormal levels of TGF-β in association with impaired neurogenesis might provide a potential mechanism for vascular abnormalities mediated defects in regenerative plasticity (Fig. 4). ...
Effect of exercise-induced neurogenesis on cognitive function deficit in a rat model of vascular dementia
1
2016
... In an animal model of VaD, a 2-vessel occlusion (2VO) in experimental rats mimicking chronic cerebral hypoperfusion (CCH) [228, 229], hippocampal neurogenesis is more severely reduced in association with cognitive impairments [230]. Physical exercise rescues the deficits in hippocampal neurogenesis and cognitive functions through the BDNF-CREB pathway in 2VO models [231]. In another study reported by Kwon et al., the inhibition of acetylcholinesterase promotes hippocampal neurogenesis and cognitive function in VaD [232]. Moreover, transplantation of MSCs provided neuroprotection in the hippocampus of 2VO models [233]. Reports on the effects of VaD induced TGF-β levels on adult neurogenesis are limited. As many animal models of AD display a vascular pathology, the regulation of adult neurogenesis by TGF-β can be expected to be similar in VaD as it is described in AD [234, 235]. In corroboration with existing findings on AD and reports generated from experimental models of VaD, it can be presumed that abnormal levels of TGF-β in association with impaired neurogenesis might provide a potential mechanism for vascular abnormalities mediated defects in regenerative plasticity (Fig. 4). ...
Effects of donepezil, an acetylcholinesterase inhibitor, on neurogenesis in a rat model of vascular dementia
1
2014
... In an animal model of VaD, a 2-vessel occlusion (2VO) in experimental rats mimicking chronic cerebral hypoperfusion (CCH) [228, 229], hippocampal neurogenesis is more severely reduced in association with cognitive impairments [230]. Physical exercise rescues the deficits in hippocampal neurogenesis and cognitive functions through the BDNF-CREB pathway in 2VO models [231]. In another study reported by Kwon et al., the inhibition of acetylcholinesterase promotes hippocampal neurogenesis and cognitive function in VaD [232]. Moreover, transplantation of MSCs provided neuroprotection in the hippocampus of 2VO models [233]. Reports on the effects of VaD induced TGF-β levels on adult neurogenesis are limited. As many animal models of AD display a vascular pathology, the regulation of adult neurogenesis by TGF-β can be expected to be similar in VaD as it is described in AD [234, 235]. In corroboration with existing findings on AD and reports generated from experimental models of VaD, it can be presumed that abnormal levels of TGF-β in association with impaired neurogenesis might provide a potential mechanism for vascular abnormalities mediated defects in regenerative plasticity (Fig. 4). ...
Transplantation of mesenchymal stem cells overexpressing interleukin-10 induces autophagy response and promotes neuroprotection in a rat model of TBI
1
2019
... In an animal model of VaD, a 2-vessel occlusion (2VO) in experimental rats mimicking chronic cerebral hypoperfusion (CCH) [228, 229], hippocampal neurogenesis is more severely reduced in association with cognitive impairments [230]. Physical exercise rescues the deficits in hippocampal neurogenesis and cognitive functions through the BDNF-CREB pathway in 2VO models [231]. In another study reported by Kwon et al., the inhibition of acetylcholinesterase promotes hippocampal neurogenesis and cognitive function in VaD [232]. Moreover, transplantation of MSCs provided neuroprotection in the hippocampus of 2VO models [233]. Reports on the effects of VaD induced TGF-β levels on adult neurogenesis are limited. As many animal models of AD display a vascular pathology, the regulation of adult neurogenesis by TGF-β can be expected to be similar in VaD as it is described in AD [234, 235]. In corroboration with existing findings on AD and reports generated from experimental models of VaD, it can be presumed that abnormal levels of TGF-β in association with impaired neurogenesis might provide a potential mechanism for vascular abnormalities mediated defects in regenerative plasticity (Fig. 4). ...
Microglia prevent peripheral immune cell invasion and promote an anti-inflammatory environment in the brain of APP-PS1 transgenic mice
3
2018
... In an animal model of VaD, a 2-vessel occlusion (2VO) in experimental rats mimicking chronic cerebral hypoperfusion (CCH) [228, 229], hippocampal neurogenesis is more severely reduced in association with cognitive impairments [230]. Physical exercise rescues the deficits in hippocampal neurogenesis and cognitive functions through the BDNF-CREB pathway in 2VO models [231]. In another study reported by Kwon et al., the inhibition of acetylcholinesterase promotes hippocampal neurogenesis and cognitive function in VaD [232]. Moreover, transplantation of MSCs provided neuroprotection in the hippocampus of 2VO models [233]. Reports on the effects of VaD induced TGF-β levels on adult neurogenesis are limited. As many animal models of AD display a vascular pathology, the regulation of adult neurogenesis by TGF-β can be expected to be similar in VaD as it is described in AD [234, 235]. In corroboration with existing findings on AD and reports generated from experimental models of VaD, it can be presumed that abnormal levels of TGF-β in association with impaired neurogenesis might provide a potential mechanism for vascular abnormalities mediated defects in regenerative plasticity (Fig. 4). ...
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... , 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Adult neurogenesis in neurodegenerative diseases
2
2015
... In an animal model of VaD, a 2-vessel occlusion (2VO) in experimental rats mimicking chronic cerebral hypoperfusion (CCH) [228, 229], hippocampal neurogenesis is more severely reduced in association with cognitive impairments [230]. Physical exercise rescues the deficits in hippocampal neurogenesis and cognitive functions through the BDNF-CREB pathway in 2VO models [231]. In another study reported by Kwon et al., the inhibition of acetylcholinesterase promotes hippocampal neurogenesis and cognitive function in VaD [232]. Moreover, transplantation of MSCs provided neuroprotection in the hippocampus of 2VO models [233]. Reports on the effects of VaD induced TGF-β levels on adult neurogenesis are limited. As many animal models of AD display a vascular pathology, the regulation of adult neurogenesis by TGF-β can be expected to be similar in VaD as it is described in AD [234, 235]. In corroboration with existing findings on AD and reports generated from experimental models of VaD, it can be presumed that abnormal levels of TGF-β in association with impaired neurogenesis might provide a potential mechanism for vascular abnormalities mediated defects in regenerative plasticity (Fig. 4). ...
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
The neuropathology and cerebrovascular mechanisms of dementia
1
2016
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Transforming growth factor-beta and ischemic brain injury
1
2003
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis
1
2009
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Cerebrospinal Fluid Inflammatory Cytokine Aberrations in Alzheimer's Disease, Parkinson's Disease and Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis
1
2018
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: An update of the Consensus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry
1
2018
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Increased hippocampal neurogenesis in Alzheimer's disease
1
2004
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Choosing an animal model for the study of Huntington's disease
1
2013
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Large Animal Models of Huntington's Disease
0
2015
Animal models of Huntington's disease
1
2007
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer's disease
1
2006
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Altered neurogenesis in mouse models of Alzheimer disease
1
2017
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Tau hyperphosphorylation affects Smad 2/3 translocation
1
2009
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Neurofibrillary tangles may interfere with Smad 2/3 signaling in neurons
0
2007
Dysfunction of TGF-beta signaling in Alzheimer's disease
2
2006
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
... ]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Alzheimer's Disease and Hippocampal Adult Neurogenesis; Exploring Shared Mechanisms
0
2016
Neurogenesis in Alzheimer's disease
1
2011
... In many neurocognitive disorders, reactive astrogliosis, microglial activation, neuroinflammation, defects in the turnover of NSCs, reactive neuroblastosis and progressive neurodegeneration of newly generated as well as existing neurons are part of the underlying pathomechanisms of the disease [58, 236]. Among them, regenerative failure resulting from abnormal neurogenesis contributes to cognitive deficits in dementia [223]. Thus, understanding the mechanism of regeneration in the adult brain and its failures has become increasingly important. Chronic high levels of TGF-β exacerbate pathomechanisms responsible for the development of dementia [38, 227, 234]. The expression of TGF-β and its downstream signaling appears to be drastically increased in the brain in response to various acute brain pathologies like ischemic stroke, brain trauma and epilepsy [213, 237, 238]. In addition, the CSF of patients with AD, Huntington’s disease (HD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), Spinocerebellar ataxia (SCA) and Multiple system atrophy (MSA) shows increased amounts of TGF-β [155, 239, 240]. Notably, increased TGF-β levels are correlated with the occurrence of reactive neurogenesis in post-mortem brain tissue derived from many of these diseases including AD, HD and ALS [241, 242]. Moreover, many neurodegenerative disorders have been characterized by stem cell quiescence and reactive neurogenesis along with elevated levels of TGF-β signaling during the early phases of pathogenesis [58, 154, 234, 235]. HD is an autosomal dominant neurodegenerative condition and is considered as a hereditary form of dementia, for which numerous experimental models are available [58, 243-245]. Among them, almost all transgenic rodent models of HD show elevated levels of TGF-β signaling in association with NSC quiescence and reactive neuroblastosis, impaired neuronal differentiation and degeneration of new neurons [58, 154, 227]. The increased level of pSmads in NSCs blocks the neurogenic process at an early stage in the hippocampus of the R6/2 mouse and tgHD rat models [154]. Besides, the induced expression of TGF-β mediated pSmad2 increases the expression of the mutant form of huntingtin, thereby leading to neurodegeneration [227]. In addition to animal models of HD, increased levels of TGF-β and a reduced level of neurogenesis were reported in genetic models of AD [61, 246, 247]. Moreover, reduced TGF-βR2 expression and defects in the phosphorylation of Smad proteins are reported to be associated with an increased level of amyloidogenesis and hyper-phosphorylation of Tau in the brain of AD subjects [248-250]. Thus, available reports on the regulation of neurogenesis in response to the abnormal levels of TGF-β appears somewhat inconsistent in AD [250-252], but this may relate to the stage of the disease and degree of the pathology. ...
Genetic reprogramming of somatic cells into neuroblasts through a co-induction of the doublecortin gene along the Yamanaka factors: A promising approach to model neuroregenerative disorders
1
2019
... Several studies have indicated that the induction of TGF-β during pathogenesis exerts a defensive role in preventing neurodegeneration [219]. Reduced expression of TGF-β and deficient Smad signaling in neurons induces neurodegeneration in the terminal stage of the disease [61]. The reduced neurogenesis seen in the brains of some models of AD could provide a possible explanation for the reduction in TGF-β mediated neurodegeneration in cognitive centers of the pathological brain. Recently, reactive neuroblasts in the diseased brain have been proposed to acquire properties of non-neuronal cells such as microglia, and thus these cells might represent a potential source of TGF-β in pathological brains [223, 253]. The robust activation, proliferation and circulation of microglia, reactive astrocytes and neuroblasts found in early pathogenesis tend to be diminished in the late phases of the diseases [227, 254]. Thus, it can be hypothesized that the overall loss of neuroblasts, non-neuronal and neuronal cells in the brain in the late phase of the disease might lead to the reduced level of TGF-β, thereby fostering neurodegeneration and memory loss. ...
Microglia in Alzheimer's Disease: Activated, Dysfunctional or Degenerative
1
2018
... Several studies have indicated that the induction of TGF-β during pathogenesis exerts a defensive role in preventing neurodegeneration [219]. Reduced expression of TGF-β and deficient Smad signaling in neurons induces neurodegeneration in the terminal stage of the disease [61]. The reduced neurogenesis seen in the brains of some models of AD could provide a possible explanation for the reduction in TGF-β mediated neurodegeneration in cognitive centers of the pathological brain. Recently, reactive neuroblasts in the diseased brain have been proposed to acquire properties of non-neuronal cells such as microglia, and thus these cells might represent a potential source of TGF-β in pathological brains [223, 253]. The robust activation, proliferation and circulation of microglia, reactive astrocytes and neuroblasts found in early pathogenesis tend to be diminished in the late phases of the diseases [227, 254]. Thus, it can be hypothesized that the overall loss of neuroblasts, non-neuronal and neuronal cells in the brain in the late phase of the disease might lead to the reduced level of TGF-β, thereby fostering neurodegeneration and memory loss. ...
The macrophage and the apoptotic cell: an innate immune interaction viewed simplistically?
1
2004
... While the potential cellular sources of the increased TGF-β levels are identified, the mechanism for the activation of TGF-β remains unknown in neurodegenerating brains. A possible interaction between dying cells and immune cells is proposed to be involved in the production of TGF-β, which in turn can mediate the inflammatory response and oxidative stress through the generation of inflammatory factors and reactive oxygen species [255, 256] (Fig. 5). Overproduction of free radicals causes oxidative damage to molecular and subcellular components of the cell, thereby leading to cell death in many tissues [257]. Oxidative stress plays a central role in inducing neurodegeneration in many dementia cases [258]. However, the effect of free radical-mediated oxidative stress on the activation of TGF-β has not been clearly established in neurocognitive disorders including VaD. While the increased level of free radicals in the vasculature and reduced bioavailability of vasodilators are associated with endothelial dysfunction leading to vascular dementia, free radicals can lead to acidosis in vascular disorders in association with the degradation of ECM [259]. As mentioned in an earlier chapter, the inactive form of TGF-β can be activated by the free-radical mediated increased acidic condition of the tissue [260]. Thus, the increased level of free radicals might be an underlying cause for triggering the activation of TGF-β in VaD. At the same time, in order to release the active form of TGF-β, the ECM needs to be degraded, which may result in the disruption of the cerebrovascular structure and breakdown of BBB. Thus, free radical-induced activation of TGF-β through the generation of acidic microenvironment could be hypothesized to play a central role in the development of VaD. Recently, Unger MS et al. provided evidence for the association of DCX-expressing T cells and microglia in the vicinity of amyloid plaques in the brain of APP-PS1 transgenic mice and human AD subjects [261]. Moreover, it could be shown that DCX-expressing immune cells exhibit indications of phagocytosis in AD brains [261]. Thus, it can be hypothesized that a potential interaction between the DCX-expressing cells and microglia may be a potential source of TGF-β during neuropathogenesis. Eventually, the elevated TGF-β might be an underlying factor for impaired neurogenesis responsible for memory loss in VaD. Taken together, TGF-β signaling is an ideal therapeutic target to intervene in pathological associated vascular defects, neurodegeneration and promote regenerative plasticity in VaD. ...
Apoptotic cells and innate immune stimuli combine to regulate macrophage cytokine secretion
1
2003
... While the potential cellular sources of the increased TGF-β levels are identified, the mechanism for the activation of TGF-β remains unknown in neurodegenerating brains. A possible interaction between dying cells and immune cells is proposed to be involved in the production of TGF-β, which in turn can mediate the inflammatory response and oxidative stress through the generation of inflammatory factors and reactive oxygen species [255, 256] (Fig. 5). Overproduction of free radicals causes oxidative damage to molecular and subcellular components of the cell, thereby leading to cell death in many tissues [257]. Oxidative stress plays a central role in inducing neurodegeneration in many dementia cases [258]. However, the effect of free radical-mediated oxidative stress on the activation of TGF-β has not been clearly established in neurocognitive disorders including VaD. While the increased level of free radicals in the vasculature and reduced bioavailability of vasodilators are associated with endothelial dysfunction leading to vascular dementia, free radicals can lead to acidosis in vascular disorders in association with the degradation of ECM [259]. As mentioned in an earlier chapter, the inactive form of TGF-β can be activated by the free-radical mediated increased acidic condition of the tissue [260]. Thus, the increased level of free radicals might be an underlying cause for triggering the activation of TGF-β in VaD. At the same time, in order to release the active form of TGF-β, the ECM needs to be degraded, which may result in the disruption of the cerebrovascular structure and breakdown of BBB. Thus, free radical-induced activation of TGF-β through the generation of acidic microenvironment could be hypothesized to play a central role in the development of VaD. Recently, Unger MS et al. provided evidence for the association of DCX-expressing T cells and microglia in the vicinity of amyloid plaques in the brain of APP-PS1 transgenic mice and human AD subjects [261]. Moreover, it could be shown that DCX-expressing immune cells exhibit indications of phagocytosis in AD brains [261]. Thus, it can be hypothesized that a potential interaction between the DCX-expressing cells and microglia may be a potential source of TGF-β during neuropathogenesis. Eventually, the elevated TGF-β might be an underlying factor for impaired neurogenesis responsible for memory loss in VaD. Taken together, TGF-β signaling is an ideal therapeutic target to intervene in pathological associated vascular defects, neurodegeneration and promote regenerative plasticity in VaD. ...
Protective Effect of Antioxidants on Neuronal Dysfunction and Plasticity in Huntington's Disease
1
2017
... While the potential cellular sources of the increased TGF-β levels are identified, the mechanism for the activation of TGF-β remains unknown in neurodegenerating brains. A possible interaction between dying cells and immune cells is proposed to be involved in the production of TGF-β, which in turn can mediate the inflammatory response and oxidative stress through the generation of inflammatory factors and reactive oxygen species [255, 256] (Fig. 5). Overproduction of free radicals causes oxidative damage to molecular and subcellular components of the cell, thereby leading to cell death in many tissues [257]. Oxidative stress plays a central role in inducing neurodegeneration in many dementia cases [258]. However, the effect of free radical-mediated oxidative stress on the activation of TGF-β has not been clearly established in neurocognitive disorders including VaD. While the increased level of free radicals in the vasculature and reduced bioavailability of vasodilators are associated with endothelial dysfunction leading to vascular dementia, free radicals can lead to acidosis in vascular disorders in association with the degradation of ECM [259]. As mentioned in an earlier chapter, the inactive form of TGF-β can be activated by the free-radical mediated increased acidic condition of the tissue [260]. Thus, the increased level of free radicals might be an underlying cause for triggering the activation of TGF-β in VaD. At the same time, in order to release the active form of TGF-β, the ECM needs to be degraded, which may result in the disruption of the cerebrovascular structure and breakdown of BBB. Thus, free radical-induced activation of TGF-β through the generation of acidic microenvironment could be hypothesized to play a central role in the development of VaD. Recently, Unger MS et al. provided evidence for the association of DCX-expressing T cells and microglia in the vicinity of amyloid plaques in the brain of APP-PS1 transgenic mice and human AD subjects [261]. Moreover, it could be shown that DCX-expressing immune cells exhibit indications of phagocytosis in AD brains [261]. Thus, it can be hypothesized that a potential interaction between the DCX-expressing cells and microglia may be a potential source of TGF-β during neuropathogenesis. Eventually, the elevated TGF-β might be an underlying factor for impaired neurogenesis responsible for memory loss in VaD. Taken together, TGF-β signaling is an ideal therapeutic target to intervene in pathological associated vascular defects, neurodegeneration and promote regenerative plasticity in VaD. ...
Oxidative stress in vascular dementia and Alzheimer's disease: a common pathology
1
2009
... While the potential cellular sources of the increased TGF-β levels are identified, the mechanism for the activation of TGF-β remains unknown in neurodegenerating brains. A possible interaction between dying cells and immune cells is proposed to be involved in the production of TGF-β, which in turn can mediate the inflammatory response and oxidative stress through the generation of inflammatory factors and reactive oxygen species [255, 256] (Fig. 5). Overproduction of free radicals causes oxidative damage to molecular and subcellular components of the cell, thereby leading to cell death in many tissues [257]. Oxidative stress plays a central role in inducing neurodegeneration in many dementia cases [258]. However, the effect of free radical-mediated oxidative stress on the activation of TGF-β has not been clearly established in neurocognitive disorders including VaD. While the increased level of free radicals in the vasculature and reduced bioavailability of vasodilators are associated with endothelial dysfunction leading to vascular dementia, free radicals can lead to acidosis in vascular disorders in association with the degradation of ECM [259]. As mentioned in an earlier chapter, the inactive form of TGF-β can be activated by the free-radical mediated increased acidic condition of the tissue [260]. Thus, the increased level of free radicals might be an underlying cause for triggering the activation of TGF-β in VaD. At the same time, in order to release the active form of TGF-β, the ECM needs to be degraded, which may result in the disruption of the cerebrovascular structure and breakdown of BBB. Thus, free radical-induced activation of TGF-β through the generation of acidic microenvironment could be hypothesized to play a central role in the development of VaD. Recently, Unger MS et al. provided evidence for the association of DCX-expressing T cells and microglia in the vicinity of amyloid plaques in the brain of APP-PS1 transgenic mice and human AD subjects [261]. Moreover, it could be shown that DCX-expressing immune cells exhibit indications of phagocytosis in AD brains [261]. Thus, it can be hypothesized that a potential interaction between the DCX-expressing cells and microglia may be a potential source of TGF-β during neuropathogenesis. Eventually, the elevated TGF-β might be an underlying factor for impaired neurogenesis responsible for memory loss in VaD. Taken together, TGF-β signaling is an ideal therapeutic target to intervene in pathological associated vascular defects, neurodegeneration and promote regenerative plasticity in VaD. ...
Free Radical Damage in Ischemia-Reperfusion Injury: An Obstacle in Acute Ischemic Stroke after Revascularization Therapy
1
2018
... While the potential cellular sources of the increased TGF-β levels are identified, the mechanism for the activation of TGF-β remains unknown in neurodegenerating brains. A possible interaction between dying cells and immune cells is proposed to be involved in the production of TGF-β, which in turn can mediate the inflammatory response and oxidative stress through the generation of inflammatory factors and reactive oxygen species [255, 256] (Fig. 5). Overproduction of free radicals causes oxidative damage to molecular and subcellular components of the cell, thereby leading to cell death in many tissues [257]. Oxidative stress plays a central role in inducing neurodegeneration in many dementia cases [258]. However, the effect of free radical-mediated oxidative stress on the activation of TGF-β has not been clearly established in neurocognitive disorders including VaD. While the increased level of free radicals in the vasculature and reduced bioavailability of vasodilators are associated with endothelial dysfunction leading to vascular dementia, free radicals can lead to acidosis in vascular disorders in association with the degradation of ECM [259]. As mentioned in an earlier chapter, the inactive form of TGF-β can be activated by the free-radical mediated increased acidic condition of the tissue [260]. Thus, the increased level of free radicals might be an underlying cause for triggering the activation of TGF-β in VaD. At the same time, in order to release the active form of TGF-β, the ECM needs to be degraded, which may result in the disruption of the cerebrovascular structure and breakdown of BBB. Thus, free radical-induced activation of TGF-β through the generation of acidic microenvironment could be hypothesized to play a central role in the development of VaD. Recently, Unger MS et al. provided evidence for the association of DCX-expressing T cells and microglia in the vicinity of amyloid plaques in the brain of APP-PS1 transgenic mice and human AD subjects [261]. Moreover, it could be shown that DCX-expressing immune cells exhibit indications of phagocytosis in AD brains [261]. Thus, it can be hypothesized that a potential interaction between the DCX-expressing cells and microglia may be a potential source of TGF-β during neuropathogenesis. Eventually, the elevated TGF-β might be an underlying factor for impaired neurogenesis responsible for memory loss in VaD. Taken together, TGF-β signaling is an ideal therapeutic target to intervene in pathological associated vascular defects, neurodegeneration and promote regenerative plasticity in VaD. ...
Acidic cellular environments: activation of latent TGF-beta and sensitization of cellular responses to TGF-beta and EGF
1
1989
... While the potential cellular sources of the increased TGF-β levels are identified, the mechanism for the activation of TGF-β remains unknown in neurodegenerating brains. A possible interaction between dying cells and immune cells is proposed to be involved in the production of TGF-β, which in turn can mediate the inflammatory response and oxidative stress through the generation of inflammatory factors and reactive oxygen species [255, 256] (Fig. 5). Overproduction of free radicals causes oxidative damage to molecular and subcellular components of the cell, thereby leading to cell death in many tissues [257]. Oxidative stress plays a central role in inducing neurodegeneration in many dementia cases [258]. However, the effect of free radical-mediated oxidative stress on the activation of TGF-β has not been clearly established in neurocognitive disorders including VaD. While the increased level of free radicals in the vasculature and reduced bioavailability of vasodilators are associated with endothelial dysfunction leading to vascular dementia, free radicals can lead to acidosis in vascular disorders in association with the degradation of ECM [259]. As mentioned in an earlier chapter, the inactive form of TGF-β can be activated by the free-radical mediated increased acidic condition of the tissue [260]. Thus, the increased level of free radicals might be an underlying cause for triggering the activation of TGF-β in VaD. At the same time, in order to release the active form of TGF-β, the ECM needs to be degraded, which may result in the disruption of the cerebrovascular structure and breakdown of BBB. Thus, free radical-induced activation of TGF-β through the generation of acidic microenvironment could be hypothesized to play a central role in the development of VaD. Recently, Unger MS et al. provided evidence for the association of DCX-expressing T cells and microglia in the vicinity of amyloid plaques in the brain of APP-PS1 transgenic mice and human AD subjects [261]. Moreover, it could be shown that DCX-expressing immune cells exhibit indications of phagocytosis in AD brains [261]. Thus, it can be hypothesized that a potential interaction between the DCX-expressing cells and microglia may be a potential source of TGF-β during neuropathogenesis. Eventually, the elevated TGF-β might be an underlying factor for impaired neurogenesis responsible for memory loss in VaD. Taken together, TGF-β signaling is an ideal therapeutic target to intervene in pathological associated vascular defects, neurodegeneration and promote regenerative plasticity in VaD. ...
Doublecortin expression in CD8+ T-cells and microglia at sites of amyloid-beta plaques: A potential role in shaping plaque pathology?
2
2018
... While the potential cellular sources of the increased TGF-β levels are identified, the mechanism for the activation of TGF-β remains unknown in neurodegenerating brains. A possible interaction between dying cells and immune cells is proposed to be involved in the production of TGF-β, which in turn can mediate the inflammatory response and oxidative stress through the generation of inflammatory factors and reactive oxygen species [255, 256] (Fig. 5). Overproduction of free radicals causes oxidative damage to molecular and subcellular components of the cell, thereby leading to cell death in many tissues [257]. Oxidative stress plays a central role in inducing neurodegeneration in many dementia cases [258]. However, the effect of free radical-mediated oxidative stress on the activation of TGF-β has not been clearly established in neurocognitive disorders including VaD. While the increased level of free radicals in the vasculature and reduced bioavailability of vasodilators are associated with endothelial dysfunction leading to vascular dementia, free radicals can lead to acidosis in vascular disorders in association with the degradation of ECM [259]. As mentioned in an earlier chapter, the inactive form of TGF-β can be activated by the free-radical mediated increased acidic condition of the tissue [260]. Thus, the increased level of free radicals might be an underlying cause for triggering the activation of TGF-β in VaD. At the same time, in order to release the active form of TGF-β, the ECM needs to be degraded, which may result in the disruption of the cerebrovascular structure and breakdown of BBB. Thus, free radical-induced activation of TGF-β through the generation of acidic microenvironment could be hypothesized to play a central role in the development of VaD. Recently, Unger MS et al. provided evidence for the association of DCX-expressing T cells and microglia in the vicinity of amyloid plaques in the brain of APP-PS1 transgenic mice and human AD subjects [261]. Moreover, it could be shown that DCX-expressing immune cells exhibit indications of phagocytosis in AD brains [261]. Thus, it can be hypothesized that a potential interaction between the DCX-expressing cells and microglia may be a potential source of TGF-β during neuropathogenesis. Eventually, the elevated TGF-β might be an underlying factor for impaired neurogenesis responsible for memory loss in VaD. Taken together, TGF-β signaling is an ideal therapeutic target to intervene in pathological associated vascular defects, neurodegeneration and promote regenerative plasticity in VaD. ...
... ]. Moreover, it could be shown that DCX-expressing immune cells exhibit indications of phagocytosis in AD brains [261]. Thus, it can be hypothesized that a potential interaction between the DCX-expressing cells and microglia may be a potential source of TGF-β during neuropathogenesis. Eventually, the elevated TGF-β might be an underlying factor for impaired neurogenesis responsible for memory loss in VaD. Taken together, TGF-β signaling is an ideal therapeutic target to intervene in pathological associated vascular defects, neurodegeneration and promote regenerative plasticity in VaD. ...
... Town T et al. reported that the inhibition of TGF-β signaling in peripheral immune cells by crossing the CD11c-DNR mice that express the CD11c promoter-driven dominant-negative TGF-β receptor type 2 with Tg2576 mice overexpressing mAPP reduced the brain parenchymal and cerebrovascular β-amyloid deposits and improved the working memory [262]. Therefore, inhibition of overactive TGF-β signaling has been proposed to attenuate neurovascular and neuro-regenerative pathologies in VaD. ...
Development and characterization of human monoclonal antibodies that neutralize multiple TGFbeta isoforms
1
2016
... TGF-β can be modulated through several distinct approaches (summarized in Figure 6). The main strategies for inhibition of TGF-β include neutralizing antibodies and recombinant fragments that block TGF-β, implementation of compounds and antisense oligonucleotides that interfere with the binding of the ligand to TGF-β receptors, and small molecules that block intracellular phosphorylation of TGF-β receptor 1 and Smad elements. Lerdelimumab (CAT-152) and metelimumab (CAT-192) are recombinant human IgGs generated by phage display technology that block TGF-β1 and TGF-β2 respectively [263]. Recombinant fusion proteins containing the ectodomains of type 2 and type 3 receptors, the targeted pan-TGF-β blocker (TTB) and soluble TGF-β II/Fc have also been used to prevent ligand binding to TGF-β receptors [264]. The synthetic agents such as SB-431542, SB-505124, SD-093, SD-908 and LY2157299 were identified to block the catalytic activity of TGF-βR1 [265]. Moreover, the antisense oligonucleotides generated against the expression of TGF-β or the TGF-β receptors can also be a promising approach to block TGF-β signaling to eventually treat VaD at an early stage of the pathogenesis. However, the safety and efficacy of these agents need to be verified in preclinical and clinical trials. Moreover, inhibition of TGF-β activity may elicit the risk for the anomalies in cell cycle regulation, cytoprotection, immunity and metabolism. Thus, unknown adverse effects of the inhibition of TGF-β related to its normal biological functions and the toxic dosages of the TGF-β agonist may not completely be excluded. Moreover, the overlapping pathophysiology between the VaD and AD needs to be dissected out, for which, the development of a valid experimental model to mimic solely the VaD that reflects the human situation needs to be accomplished. However, further studies are needed to appraise the discussed information and proposed ideas of this article for the most relevant clinical information. ...
Ligand-dependent and -independent interactions with the transforming growth factor type II and I receptor subunits reside in the aminoterminal portion of the ectodomain of the type III subunit
1
1998
... TGF-β can be modulated through several distinct approaches (summarized in Figure 6). The main strategies for inhibition of TGF-β include neutralizing antibodies and recombinant fragments that block TGF-β, implementation of compounds and antisense oligonucleotides that interfere with the binding of the ligand to TGF-β receptors, and small molecules that block intracellular phosphorylation of TGF-β receptor 1 and Smad elements. Lerdelimumab (CAT-152) and metelimumab (CAT-192) are recombinant human IgGs generated by phage display technology that block TGF-β1 and TGF-β2 respectively [263]. Recombinant fusion proteins containing the ectodomains of type 2 and type 3 receptors, the targeted pan-TGF-β blocker (TTB) and soluble TGF-β II/Fc have also been used to prevent ligand binding to TGF-β receptors [264]. The synthetic agents such as SB-431542, SB-505124, SD-093, SD-908 and LY2157299 were identified to block the catalytic activity of TGF-βR1 [265]. Moreover, the antisense oligonucleotides generated against the expression of TGF-β or the TGF-β receptors can also be a promising approach to block TGF-β signaling to eventually treat VaD at an early stage of the pathogenesis. However, the safety and efficacy of these agents need to be verified in preclinical and clinical trials. Moreover, inhibition of TGF-β activity may elicit the risk for the anomalies in cell cycle regulation, cytoprotection, immunity and metabolism. Thus, unknown adverse effects of the inhibition of TGF-β related to its normal biological functions and the toxic dosages of the TGF-β agonist may not completely be excluded. Moreover, the overlapping pathophysiology between the VaD and AD needs to be dissected out, for which, the development of a valid experimental model to mimic solely the VaD that reflects the human situation needs to be accomplished. However, further studies are needed to appraise the discussed information and proposed ideas of this article for the most relevant clinical information. ...
Targeting the transforming growth factor-beta signaling pathway in human cancer
1
2010
... TGF-β can be modulated through several distinct approaches (summarized in Figure 6). The main strategies for inhibition of TGF-β include neutralizing antibodies and recombinant fragments that block TGF-β, implementation of compounds and antisense oligonucleotides that interfere with the binding of the ligand to TGF-β receptors, and small molecules that block intracellular phosphorylation of TGF-β receptor 1 and Smad elements. Lerdelimumab (CAT-152) and metelimumab (CAT-192) are recombinant human IgGs generated by phage display technology that block TGF-β1 and TGF-β2 respectively [263]. Recombinant fusion proteins containing the ectodomains of type 2 and type 3 receptors, the targeted pan-TGF-β blocker (TTB) and soluble TGF-β II/Fc have also been used to prevent ligand binding to TGF-β receptors [264]. The synthetic agents such as SB-431542, SB-505124, SD-093, SD-908 and LY2157299 were identified to block the catalytic activity of TGF-βR1 [265]. Moreover, the antisense oligonucleotides generated against the expression of TGF-β or the TGF-β receptors can also be a promising approach to block TGF-β signaling to eventually treat VaD at an early stage of the pathogenesis. However, the safety and efficacy of these agents need to be verified in preclinical and clinical trials. Moreover, inhibition of TGF-β activity may elicit the risk for the anomalies in cell cycle regulation, cytoprotection, immunity and metabolism. Thus, unknown adverse effects of the inhibition of TGF-β related to its normal biological functions and the toxic dosages of the TGF-β agonist may not completely be excluded. Moreover, the overlapping pathophysiology between the VaD and AD needs to be dissected out, for which, the development of a valid experimental model to mimic solely the VaD that reflects the human situation needs to be accomplished. However, further studies are needed to appraise the discussed information and proposed ideas of this article for the most relevant clinical information. ...
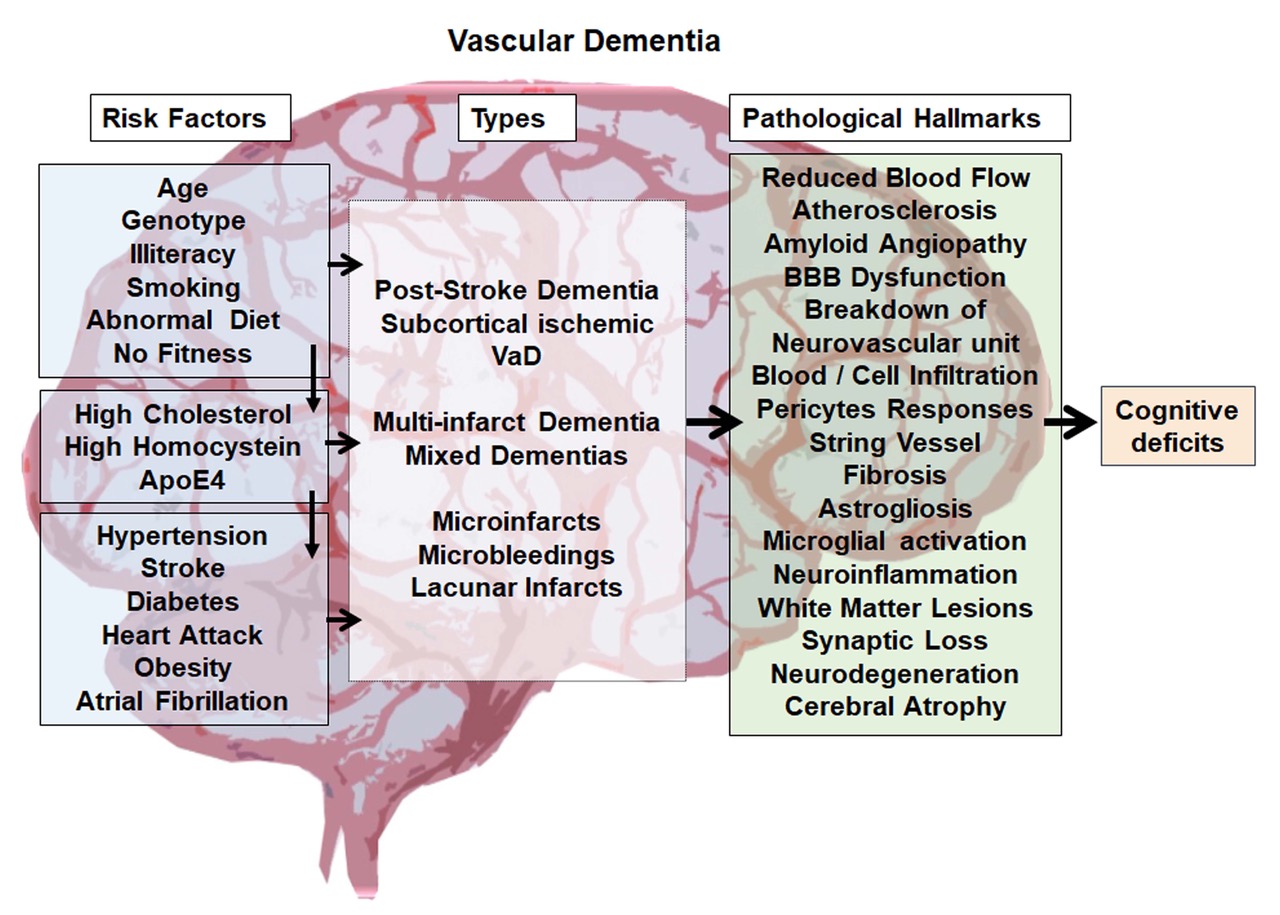
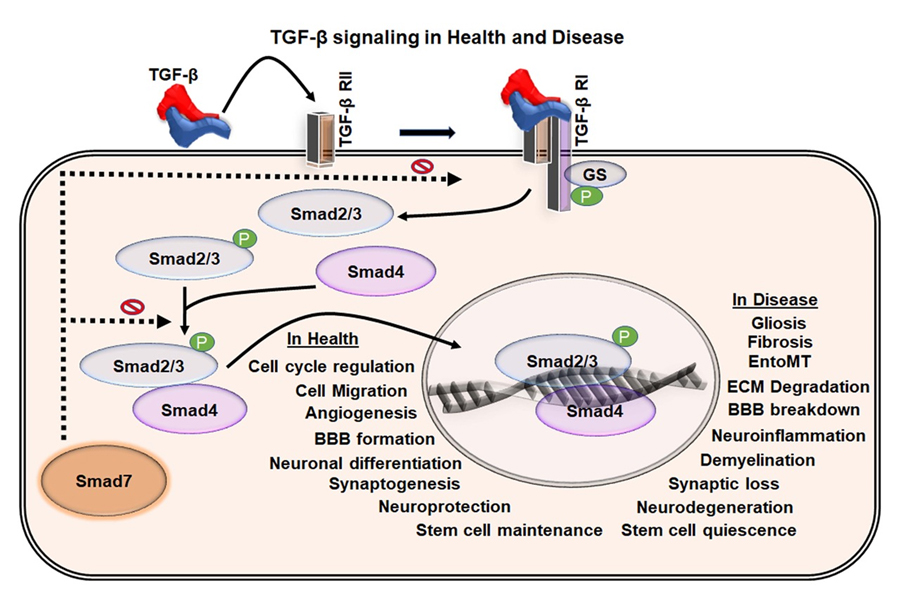
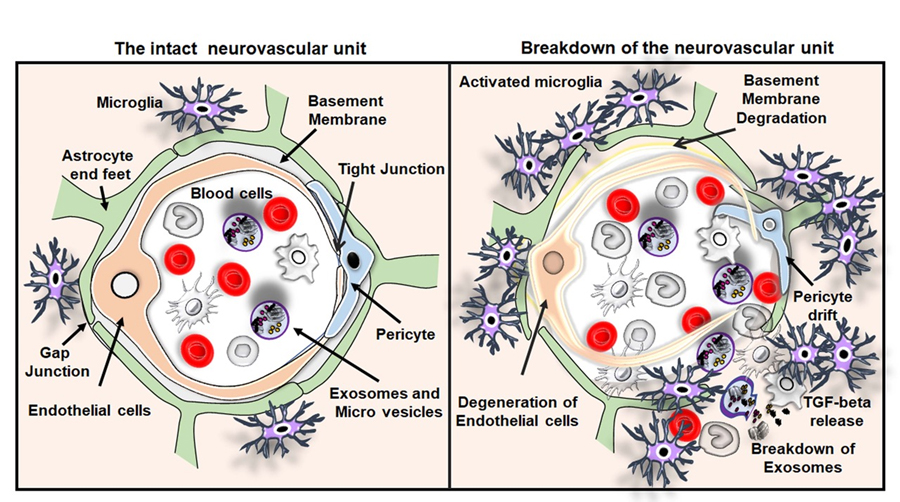
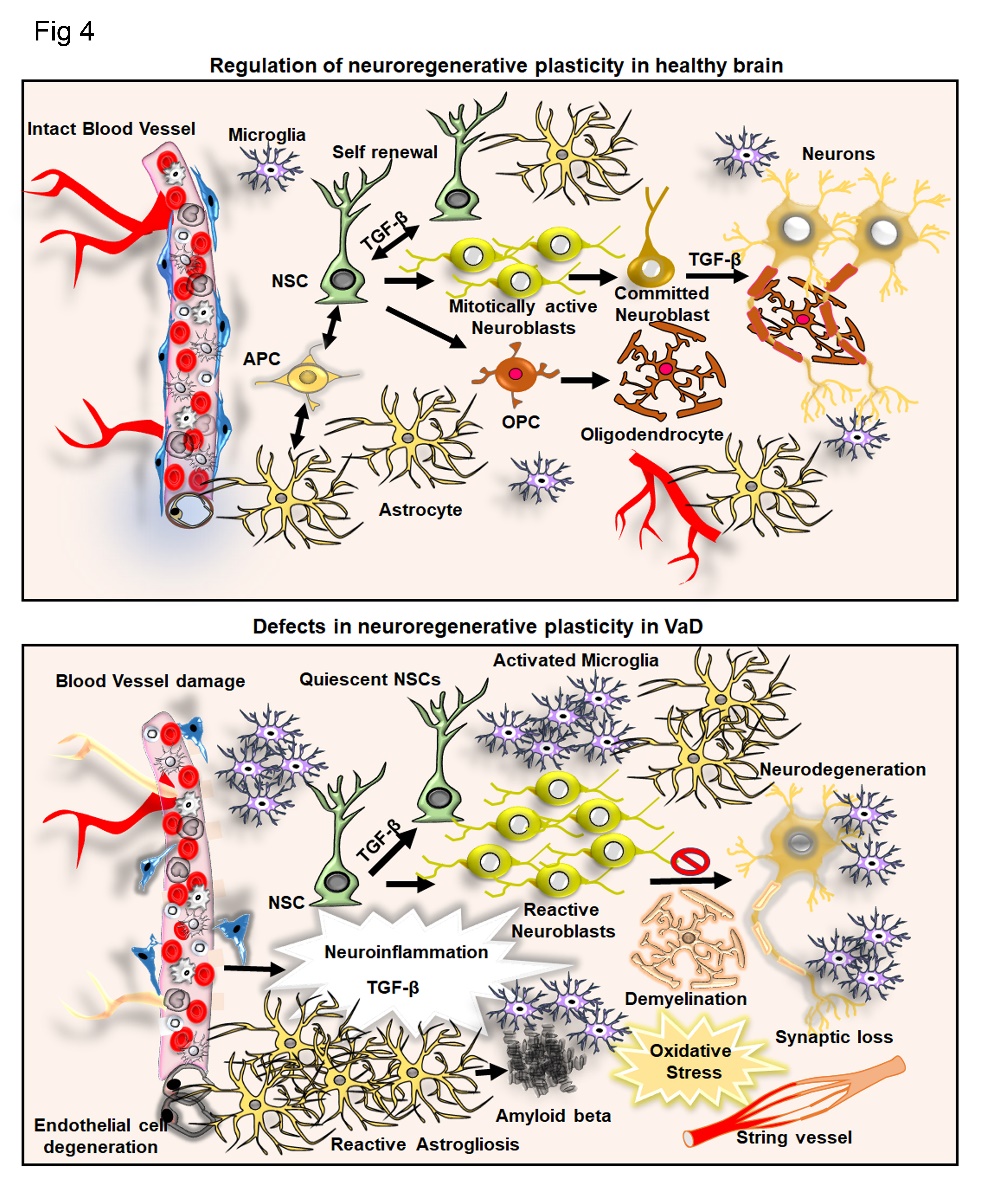
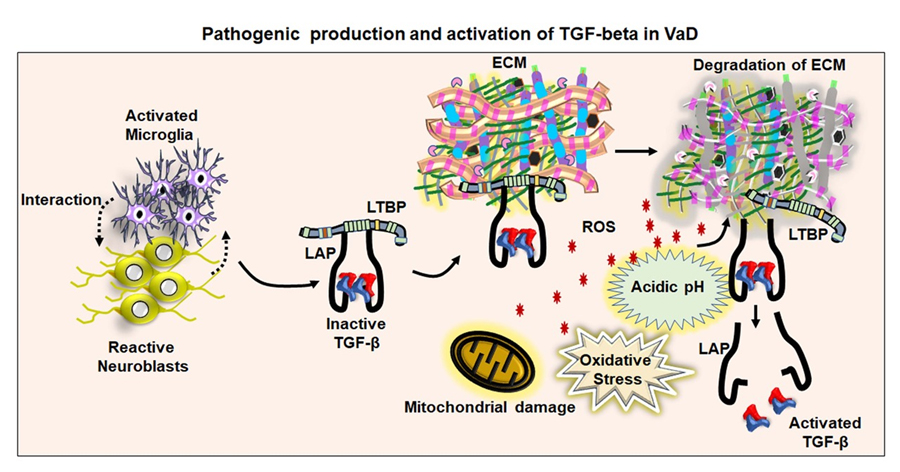
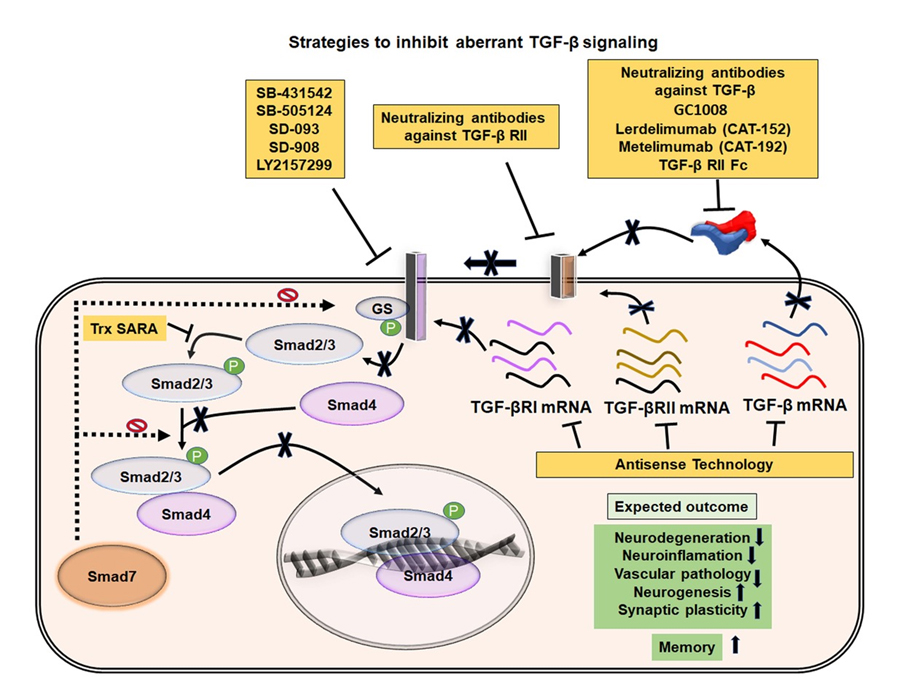
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



