An Senbo1, Hu Huiyu2, Li Yusheng1, 3, *, Hu Yihe1, *
1Department of Orthopedics, Xiangya Hospital, Central South University, Changsha, Hunan, China.2Department of General Surgery, Xiangya Hospital, Central South University, Changsha, Hunan, China.3National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, Hunan, China.
通讯作者: Correspondence should be addressed to: Drs. Yusheng Li and Yihe Hu, Department of Orthopedics, Xiangya Hospital, Central South University, Changsha, Hunan, China. E-mail: liyusheng@csu.edu.cn (Y.L) and huyh1964@163.com (Y. H).Correspondence should be addressed to: Drs. Yusheng Li and Yihe Hu, Department of Orthopedics, Xiangya Hospital, Central South University, Changsha, Hunan, China. E-mail: liyusheng@csu.edu.cn (Y.L) and huyh1964@163.com (Y. H).Correspondence should be addressed to: Drs. Yusheng Li and Yihe Hu, Department of Orthopedics, Xiangya Hospital, Central South University, Changsha, Hunan, China. E-mail: liyusheng@csu.edu.cn (Y.L) and huyh1964@163.com (Y. H).
收稿日期:2019-08-19
修回日期: 2019-11-9
接受日期: 2019-11-27
网络出版日期: 2020-10-01
版权声明:
2020 this is an open access article distributed under the terms of the creative commons attribution license, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
展开
Abstract
Recent studies have revealed novel forms of cell death beyond the canonical types of cellular apoptosis and necrosis, and these novel forms of cell death are induced by extreme microenvironmental factors. Pyroptosis, a type of regulated cell death, occurs when pattern recognition receptors (PRRs) induce the activation of cysteine-aspartic protease 1 (caspase-1) or caspase-11, which can trigger the release of the pyrogenic cytokines interleukin-1β (IL-1β) and IL-18. Osteoarthritis (OA), the most common joint disease worldwide, is characterized by low-grade inflammation and increased levels of cytokines, including IL-1β and IL-18. Additionally, some damaged chondrocytes associated with OA exhibit morphological changes consistent with pyroptosis, suggesting that this form of regulated cell death may contribute significantly to the pathology of OA. This review summarizes the molecular mechanisms of pyroptosis and shows the critical role of NLRP3 (NLR family, pyrin domain containing 3; NLR refers to “nucleotide-binding domain, leucine-rich repeat”) inflammasomes. We also provide evidence describing potential role of pyroptosis in OA, including the relationship with OA risk factors and the contribution to cartilage degradation, synovitis and OA pain.
AnSenbo, HuHuiyu, LiYusheng, HuYihe. Pyroptosis Plays a Role in Osteoarthritis[J]. Aging and Disease, 2020, 11(5): 1146-1157 https://doi.org/10.14336/AD.2019.1127
1.Introduction
Cell death is one of the most fundamental physiological processes used by multicellular organisms to maintain homeostasis, as exemplified by tissue sculpting during embryologic development and the elimination of senescent cells. Cell death is also a characteristic of abnormal pathological responses to harmful stimuli, such as physical damage and pathogens [1]. Traditionally, the literature concerning cell death has been based on the morphology of dying cells and is dominated by apoptosis and necrosis [2]. Apoptosis, a process driven by the caspase family of enzymes, is the most studied and best characterized form of programmed cell death [3]. Morphological changes observed in apoptotic cells include DNA fragmentation, decreased cell volume and cytoplasmic and membrane vesicle formation [4]. Ultimately, apoptotic cells are cleared rapidly by adjacent phagocytes without inducing an inflammatory response. Because of these characteristics, apoptosis is considered to be “programmed” and “regulated” and is beneficial for normal embryonic development, tissue homeostasis and immune responses [5]. In contrast, necrosis is described as an uncontrolled and “accidental” process caused by exposure to overwhelming microenvironmental conditions such as extreme hyperthermia, hypothermia or radiation [6]. Necrotic cells exhibit swelling, plasma membrane rupture and increased permeability, as well as a loss of cellular contents and maintenance of DNA contents [7]. Necrosis is a strongly inflammatory process with little phagocytic involvement.
Recent studies have revealed several other types of cell death, including autophagy [8], necroptosis [9], pyroptosis [10], ferroptosis [11], mitochondrial permeability transition (MPT)-dependent necrosis [12] and parthanatos [13]. These discoveries have led to the concept that multiple forms of regulated cell death can encompass morphological and biochemical features similar to both apoptosis and necrosis [14]. Pyroptosis is a type of proinflammatory programmed cell death that is triggered by inflammasomes. This process induces cell rupture and the release of cell contents, similar to necrosis, but pyroptosis involves caspase-driven programmed cell death, which is consistent with apoptosis [15].
Osteoarthritis (OA) is the most common chronic degenerative disease worldwide. Although OA is highly prevalent [16], it mainly affects elderly people. This disease can involve nearly any joint in the human body, and the most common symptoms include joint pain and disordered articular functions [17]. The pathological changes associated with OA affect all tissues in the joint and include cartilage degeneration, subchondral sclerosis, variable degrees of synovial inflammation, osteophyte formation and hypertrophy of the whole joint capsule [18].
Inflammasomes such as NLRP3 (NLR family, pyrin domain containing 3; NLR refers to “nucleotide-binding domain, leucine-rich repeat”), which are induced by nuclear factor kappa B (NF-κB) signaling and can convert interleukin-1β (IL-1β) and IL-18 into mature proinflammatory cytokines, are considered a factor in low-grade inflammatory pathology [19]. Among these inflammasomes, which comprise NLRs (NLRP1, NLRP3, and NLRC4), absent in melanoma 2 (AIM2), IFN-inducible protein 16 (IFI16) and pyrin, the NLRP3 inflammasome is the best characterized. This complex comprises NLRP3, apoptosis-related speck-like protein containing (ASC) a caspase recruitment domain (CARD) and procaspase-1 [20], suggesting that pyroptosis may be closely involved in the pathological changes associated with OA. Furthermore, the frequency of OA pain was indicated to be mediated by aberrantly overexpressed cytokines and to be associated strongly with inflammatory severity, especially in soft tissues such as the synovium, which could lead to hypersensitivity with exaggerated pain (hyperalgesia) in response to noxious stimuli or innocuous stimuli that are perceived as painful (allodynia) [21]. This indicates that pyroptosis may contribute to OA pain through releasing related cytokines [22]. In this review, we aimed to identify the possible mechanism by which pyroptosis contributes to OA progression and pain. Because no efficient treatment option is currently available for OA, an understanding of pyroptosis and related signaling factors might be useful in clinical decision-making.
2.Pyroptosis
Classic apoptosis is a caspase-dependent process of programmed cell death. An alternative form of cell death involving caspase-dependent programmed cell death combined with proinflammatory changes has also been identified [23]. This process was termed “pyroptosis” [24] after the identification of macrophages infected with Salmonella or Shigella spp. The term for this type of proinflammatory programmed cell death was derived from the Greek roots pyro, meaning “fire” or “fever,” and ptosis, which means “falling” [25]. In contrast to other forms of programmed cell death, pyroptosis is closely related to inflammation. Morphologically, pyroptosis involves the cleavage of procaspase-1 to yield active caspase-1. This activated enzyme forms pores in the plasma membrane, which enables the equalization of the ionic gradient between the intracellular and extracellular environments. Consequently, water flows into the cell, causing swelling and lysis [26]. Along with the increase in cell size and formation of a balloon-like surrounding vesicle, the nucleus condenses and becomes rounded [27, 28]. Although DNA fragmentation consistent with apoptosis is observed, nuclear integrity is maintained in pyroptosis [29, 30]. Therefore, the morphological changes associated with pyroptosis are distinct from those associated with apoptosis, and the former process can be described as having a combination of apoptotic and necrotic features.
3.Molecular mechanisms of pyroptosis
Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47].
Noncanonical inflammasomes detect intracellular bacteria and bacterial lipopolysaccharide (LPS) molecules in infected cells [48] and consequently activate caspase-11 (in mice) or caspase-4/5 (in humans). Subsequently, caspase-11 directly triggers pyroptosis. Once activated, these caspases cleave the cellular protein GSDMD, which inserts into the cell membrane lipid bilayer via its N-terminal domain. This process enables the formation of pores that induce osmotic cell lysis [49, 50]. A previous study proved that caspase-11 also indirectly processes IL-1β via the caspase-1 pathway [37], which suggests crosstalk between different caspases during the pyroptosis.
4.Pyroptosis in different diseases
Several studies have investigated the roles of pyroptosis in different diseases, particularly cardiovascular diseases[51]. Existing evidence has revealed significant increases in caspase-1 expression in vulnerable plaques and ruptured lesions associated with acute coronary events in atherosclerosis [52]. Additionally, monocyte/ macrophage pyroptosis may also induce an amplified inflammatory response and promote plaque rupture and thrombosis, which can induce acute cardiovascular events [53]. Moreover, vascular smooth muscle cell pyroptosis may suppress the healing of vascular injuries and reduce the stability of atherosclerotic plaques [54, 55]. Cardiomyocyte and cardiac fibroblast pyroptosis is also involved in diabetic cardiomyopathy [56], cardiac hypertrophy [57] and ischemic heart disease [58].
Existing evidence suggests that inflammasomes such as NLRP3 can be activated by fatty acids and high glucose levels, leading to chronic intestinal inflammation [59] and colorectal cancer [60]. These findings indicate that pyroptosis plays a significant role in digestive tract inflammation as well as in obesity-associated carcinogenesis. Pyroptosis, a highly inflammatory mode of cell death, can also trigger autoimmune diseases such as systemic lupus erythematosus (SLE) [61]. LPS from gram-negative bacterial infection induce pyroptosis by activating caspase-4/5/11, contributing to infectious diseases such as sepsis [25]. Pyroptosis also plays a role in inflammatory joint diseases such as rheumatoid arthritis (RA) [62] and OA [19].
5.Osteoarthritis
As noted previously, OA is the most common degenerative joint disorder and can affect nearly any small or large joint in the body [63]. OA is now considered to have complex etiology, and these multiple factors can lead to similar outcomes of joint destruction and clinical features [64]. In a study of OA patients older than 50 years of age in North America and Europe, approximately 60%, 33% and 5% reported involvement of joints of the hand, knee and hip, respectively [65]. An age older than 40 years is associated with a greater likelihood of developing OA. Moreover, OA is more frequent in women than in men at any age older than 50 years (i.e., postmenopausal women), and this sex difference mainly involves the hand (e.g., distal interphalangeal) and knee joints [66]. The risk factors for OA include factors related to individual susceptibility, as well as those affecting biomechanical joint stability [67]. Obesity may also be a risk factor for OA, as it increases stress on weight-bearing joints such as the hips and knees. Additionally, fat cells produce potentially harmful inflammatory proteins that may target the joints. The genetics underlying OA are complex but highly significant and may be related to changes in important molecular pathways. The genes encoding inflammatory and catabolism-related proteins are upregulated during OA, primarily through signal transduction involving nuclear factor-κB (NF-κB), mitogen- activated protein kinase, and other inflammation- and stress-induced pathways [63, 68]. Joint injuries, such as those involving the ligaments and meniscus, can increase the fragility of the joint and increase the risk of osteoarthritis. Even injuries that occurred many years ago can increase the risk of OA. Moreover, joint malformation or cartilage defects can increase the risk of OA [69-71]. The symptoms of OA include pain, transient morning stiffness and a grating sensation when walking. End-stage OA is characterized by instability and physical disability, which have tremendous negative effects on the quality of life [72]. The lack of efficient and curative treatment options for OA and the ever-evolving pathophysiological and risk factors present significant challenges.
Because its exact pathogenesis has not been fully revealed, OA is characterized as a whole-joint disease associated with pathological changes in all involved tissues. The pathology of OA includes the progressive loss and destruction of cartilage, sclerosis of the subchondral bone, formation of osteophytes, variable degrees of synovial inflammation, degeneration of the ligaments and meniscus and hypertrophy of the whole joint capsule[73, 74]. Initially, the risk factors that cause joint instability, such as ligament injury and excessive body weight, induce the excessive secretion of transforming growth factor β1 (TGF-β1) in the subchondral bone. An excess of this cytokine leads to the uncoupling of bone formation and resorption, which is accompanied by angiogenesis, sensory innervation [21], bone cavity formation and sclerosis [75]. Articular cartilage degeneration is the primary concern in OA [76]. Normally, in an environment with low oxygen, chondrocytes exhibit a low-grade turnover rate to maintain an anabolic-catabolic balance in the cartilage [77]. In OA, however, the expression of genes that encode proteins associated with inflammatory and catabolic responses is upregulated primarily via signal transduction pathways involving NF-κB, mitogen-activated protein kinase and other factors that are activated by inflammation. This transcriptional activity increases the production of primary inflammatory cytokines, such as IL-1β and tumor necrosis factor α (TNF-α), which play critical roles in OA pathology. IL-1β and TNF-α stimulate chondrocytes to release cartilage-degrading enzymes such as metalloproteinases 1, 3 and 13 (MMP1, MMP3 and MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 [78, 79]. These enzymes digest aggrecans and type II collagen to degrade the cartilage matrix. IL-1β and TNF-α also contribute to inflammatory chondrocyte death [80]. Together, these processes lead to a gradual loss of cartilage and the formation of osteochondral fissures, which allow the formation of new endochondral bone accompanied by angiogenesis and sensory innervation [81]. These processes may be closely associated with OA pain. During the late stage of OA, patients lose joint function and present with an immensely narrowed joint space containing little remaining cartilage, especially in the knee and hip joints.
The synovium is an important source of nutrients for cartilaginous tissue, which is avascular. Therefore, synovitis, or inflammation of the synovium, plays a key role in OA pathogenesis [82]. The synovium includes the synovial membrane and fluid [83]. Damaged chondrocytes and cartilage release proteinases that increase the concentrations of proinflammatory cartilage degradation products, such as DAMPs, in the synovial fluid. These products induce inflammation in the adjacent synovium [84, 85] and present feedback to the chondrocytes for further degradation. Consequently, increased amounts of matrix-degrading enzymes are secreted to deteriorate the cartilage, and reduced amounts of lubricin and hyaluronic acid (HA) are secreted to protect the joints [86].
6.The role of pyroptosis in OA
6.1Pyroptosis is related to OA risk factors
To date, a limited amount of literature has described the role of the NLRP3 inflammasome in OA pathogenesis. Since strong evidence shows that NLRP3 is a potential marker in other diseases such as RA [87], atherosclerosis [88], gout [89] and colorectal cancer [90], we detailed the evidence that indicates the association of NLRP3 with OA in the following sections.
Cartilage degeneration is the most well-known pathological change associated with OA. The joint tissues of patients harboring OA risk factors, such as metabolic disorders, aging, infectious joint diseases and injuries, can generate several kinds of DAMPs or PAMPs, including adipokines, microcrystals and uric acid.
Metabolic disorders contribute to OA pathogenesis. Obesity (body mass index > 30 kg/m2) is a significant risk factor for the onset and progression of OA [91]. Waist circumference, which is an indicator of central adiposity, is associated with increased knee cartilage defects [92], reduced knee cartilage mass and increased bone marrow lesions [93]. Adipokines, one of the obesity-related metabolic factors, have been shown to induce cartilage degradation [64]. Leptin, an adipokine, has a detrimental effect on chondrocyte proliferation by increasing IL-1β and MMPs [94]. Additionally, individuals with obesity have low circulating 25-hydroxy-vitamin D (25-(OH)D) [95], which is positively associated with the development and worsening of knee OA, including cartilage loss and increased OA pain [96].
Adipose tissue macrophages (ATMs) exhibit a proinflammatory M1 phenotype under obese conditions but an M2-dominant phenotype under normal conditions. Proinflammatory ATMs produce tumor-promoting cytokines, including TNF-α, IL-1β, IL-6, IL-8, IL-18 and IL-32 [95], and therefore serve as a source of both local and systemic proinflammatory mediators [96]. Factors such as oxidized low-density lipoprotein (LDL) and cholesterol can activate the NLRP3 inflammasome. Vandanmagsar et al [97] found that ablation of NLRP3 in mice prevented obesity-induced inflammasome activation in fat deposits. In other words, obesity promotes pyroptosis by enabling the release of the NLRP3 inflammasome, which may be related to OA progression. Furthermore, humans with obesity have elevated blood LPS levels. LPS activates caspase-4/5/11, which initiates pyroptosis via the noncanonical inflammasome pathway [99] at both the systemic and tissue levels, leading to increased inflammatory chondrocyte death and loss of cartilage.
Microcrystals, including basic calcium phosphate (BCP), calcium pyrophosphate and uric acid [98], are considered “danger signals” and are thought to be the main DAMPs that activate inflammasomes. BCP crystals have been found in the knee and hip joints of OA patients [99], and these crystals elicit the release of IL-1β from macrophages by activating NLRP3 in a process that is dependent on K+ efflux, reactive oxygen species (ROS) and phagocytosis [100, 101]. This pyroptosis-related process drives the pathology of OA and induces cartilage degeneration. Previous reports[102] suggested that in synovial fibroblasts and articular chondrocytes, IL-36, an IL-1 cytokine subfamily member, stimulates the expression of inflammatory mediators and thus plays a critical role in inflammatory diseases [59].
Aging is also a significant risk factor for multiple chronic diseases, as elderly people are more vulnerable due to their decreased immune and metabolic activities. A highly critical event occurs with aging, which starts with DAMP accumulation and activation of the subsequent NLRP3 inflammasome, which is stimulated by endogenous byproducts [103] that are then recognized by PRRs in macrophages to trigger chronic, low-grade inflammation [31]. In turn, chronic inflammation plays an important role in the progression of metabolic disorders, such as obesity, gouty arthritis and atherosclerosis [104].
6.2Pyroptosis contributes to cartilage degradation
Upon stimulation by DAMPs, macrophages surrounding the cartilage undergo pyroptosis after caspase-1 activation by DAMPs or PAMPs and the release inflammasomes such as NLRP3. This increases the release of IL-1β and IL-18 on the cartilage surface, leading to increases in the concentrations of proinflammatory cytokines in chondrocytes and further promoting pyroptosis [105]. These cytokines stimulate chondrocytes to secrete catabolic enzymes that cause cartilage degradation, such as MMP13 and ADAMTS5 [106], thus leading to cartilage destruction.
6.3Pyroptosis plays a key role in synovial changes in OA
Chronic synovial inflammation is a known cause of cartilage degradation [87, 107]. In OA, IL-1β, which is involved in cartilage degradation, may be produced by synovial tissue rather than by chondrocytes [108]. Furthermore, the protein level of NLRP3 is more than five-fold higher in the synovial membranes of OA patients than in subjects with normal joints [109]. Genetic studies have proven a close association of inflammasomes with RA, which is a primary autoimmune disease of the synovium. These studies revealed the significantly upregulated expression of genes encoding NLRP3 inflammasome components, including NLRP3, ASC and caspase-1, in patients with RA [62, 110].
In patients with knee OA, the level of uric acid, a danger signal, in the synovial fluid was found to correlate with the synovial levels of IL-1β and IL-18 and the severity of OA [111]. Therefore, uric acid is a driver of OA pathogenesis and may be a marker of OA severity [112]. Chondrocytes produce increased IL-1β and IL-18, while IL-1β also independently induces the expression of IL-18, which enhances the local inflammatory activities independently in the surrounding synovial fluid [113], enhancing local inflammation during the progression of OA.
Briefly, in OA synovial macrophages, pyroptosis-associated inflammasomes, such as NLRP3, are induced by different DAMPs and released into the synovial fluid. The concentrations of these inflammasomes are elevated in the articular fluid and surrounding tissues. They increase the level of IL-1β and stimulate chondrocytes to produce more proinflammatory cytokines (e.g., IL-18) in a cascade of inflammatory responses involving macrophages and chondrocytes. Ultimately, this process leads to chondrocyte pyroptosis and cartilage degradation.
6.4Pyroptosis contributes to OA pain
Pyroptosis may also contribute to the pathological mechanism underlying pain, which is one of the most characteristic symptoms of OA. Continuous nociceptive input from the OA joint is thought to sensitize central and peripheral nerves and induce pain [114]. The majority of OA pain is caused by changes in peripheral nerves. Normally, nerve nociceptors are localized within specific tissues and can only detect sensations in a designated area. During OA progression, excess cytokines, chemokines and inflammatory factors, mechanical stimuli and enhanced innervation contribute to the expansion of nociceptive input [115]. In OA pathology, inflammasomes in pyroptotic macrophages upregulate the production of proinflammatory cytokines, such as IL-1β, IL-18 and TNF-α, which can increase nociceptive input in the joint tissue. This process is called “hyperalgesia.” Additionally, in the context of chronic inflammation associated with OA, pyroptosis may contribute to pathological changes in the whole joint, including the loss of cartilage, formation of osteochondral fissures and osteophytes and synovitis [116]. Furthermore, dense networks of perivascular sensory and sympathetic nerves may innervate the channels or extend into the synovium, ligament or meniscus [116, 117]. This abnormal pattern of innervation would surely contribute to OA pain.
Figure 1. Pyroptosis in knee osteoarthritis progression. In macrophages or other cells in the synovium or synovial fluid of the knee joint, DAMPs can stimulate and combine with PRRs (e.g., TLRs) on the membrane, forming inflammasomes (ASC, pro-caspase-1 and NLRP3), which are activated by two steps: priming and activation. Inflammasomes can convert pro-caspase-1 into active caspase-1, which cleaves GSDMD and form GSDMD-N. GSDMD-N generates membrane pores and leads to cell swelling and eventual lysis. Moreover, caspase-1 induces the release of more mature proinflammatory cytokines, including IL-1β and IL-18, which can in turn lead to further inflammatory cytokine release and promote pyroptosis. This is the canonical pathway of pyroptosis. LPS can directly activate caspase-4/5 (human) or caspase-11 (mouse), which can also activate GSDMD, and this is a noncanonical pyroptosis pathway. Compared with the inflammatory activity in normal knee joints, the increased inflammatory activities caused by pyroptosis in the joint promote osteoarthritis progression, with exacerbated synovitis, cartilage degradation and increased sensory input to the central nervous system, which can generate more pain.
7.Conclusions and perspectives
Pyroptosis is a proinflammatory form of regulated cell death that may be strongly correlated with OA progression. The levels of DAMPs, such as microcrystals, are much higher in OA joints and can trigger activation of the NLRP3 inflammasome and caspase-1. Much higher levels of NLRP3 are found in the synovium in OA joints than in normal joints, suggesting that macrophage pyroptosis is closely related to synovial inflammation. Pyroptotic cells release IL-1β and IL-18 into the synovial fluid following the activation of caspase-1 (i.e., the canonical pyroptosis pathway). This process further increases the levels of proinflammatory cytokines in chondrocytes and enhances the inflammatory response cascade, ultimately leading to chondrocyte death and cartilage degradation. Additionally, in patients with obesity, elevated LPS levels directly trigger the noncanonical pyroptosis pathway by activating caspase-4/5. Moreover, the proinflammatory cytokines generated during pyroptosis contribute to an increase in nociceptive input in the joint, which can cause a state of hyperalgesia and OA pain. Additionally, destruction of the cartilage and other joint tissues enables excessive sensory and sympathetic innervation, which further exaggerates the sensation of pain. The molecular mechanism underlying pyroptosis is well recognized. Accordingly, the inhibition of inflammasomes such as NLRP3, caspase-1 or GSDMD may represent a novel approach to the treatment of OA.
However, our understanding of the role of pyroptosis in OA remains limited. Moreover, there were also some opposing data that do not support the role of pyroptosis in OA. Busso et al [118] found in a murine model that IL-1α and IL-1β were not key mediators in OA. A lack of IL-1α or NLRP3 could lead to increased cartilage erosion. Bougault et al [111] found that knockout of NLRP3 did not inhibit the expression of MMP3, MMP9 and MMP13 in cultured chondrocytes, and this was no changed by inhibiting caspase-1 or IL-1β. These studies all indicated that inhibiting IL-1β was not a promising therapeutic target, which was consistent with human clinical trials[119]. However, these data do not necessarily disprove the role of pyroptosis in OA. First, the animal and cartilage explant models in these studies may not accurately reflect the aspects of the disease that are mediated by pyroptosis. Second, pyroptosis may be a process that is closely related to cell death, not just inflammatory factors such as IL-1β.
Further studies should focus on the mechanism of pyroptosis in chondrocytes and other nonmacrophage cells to clarify how this process of cell death is mediated in chondrocytes within cartilaginous tissues. This knowledge would improve our understanding of the mechanism by which pyroptosis mediates OA development and could elucidate the mechanism underlying OA pathogenesis. Overall, the role of inflammasomes such as NLRP3 and their regulators in pyroptosis indicates that NLRP3 appears to be a promising biomarker for the diagnosis and monitoring of OA. Treatment targeting NLRP3 may be a potential therapeutic strategy for OA.
Acknowledgments
This review is supported by several fundings: National Key R&D Program of China (grant number: 2019 YFA0111900), National Natural Science Foundation of China (NSFC) projects (grant numbers: 81873988, 81874030 and 81601883) and supported by the Fundamental Research Fund for the Central Universities of Central South University (grant number: 2018zzts257).
Conflicts of interest
The authors declare that they have no conflicts of interest pertaining to this study.
CourbetA, BecN, ConstantC, LarroqueC, PugniereM, El MessaoudiS, et al. (2017).
Imidazoquinoxaline anticancer derivatives and imiquimod interact with tubulin: Characterization of molecular microtubule inhibiting mechanisms in correlation with cytotoxicity
MathewsRJ, RobinsonJI, BattellinoM, WongC, TaylorJC, EyreS, et al. (2014).
Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment
Revisiting the Role of Interleukin-1 Pathway in Osteoarthritis: Interleukin-1alpha and -1beta, and NLRP3 Inflammasome Are Not Involved in the Pathological Features of the Murine Menisectomy Model of Osteoarthritis
Die another way--non-apoptotic mechanisms of cell death
1
2014
... Cell death is one of the most fundamental physiological processes used by multicellular organisms to maintain homeostasis, as exemplified by tissue sculpting during embryologic development and the elimination of senescent cells. Cell death is also a characteristic of abnormal pathological responses to harmful stimuli, such as physical damage and pathogens [1]. Traditionally, the literature concerning cell death has been based on the morphology of dying cells and is dominated by apoptosis and necrosis [2]. Apoptosis, a process driven by the caspase family of enzymes, is the most studied and best characterized form of programmed cell death [3]. Morphological changes observed in apoptotic cells include DNA fragmentation, decreased cell volume and cytoplasmic and membrane vesicle formation [4]. Ultimately, apoptotic cells are cleared rapidly by adjacent phagocytes without inducing an inflammatory response. Because of these characteristics, apoptosis is considered to be “programmed” and “regulated” and is beneficial for normal embryonic development, tissue homeostasis and immune responses [5]. In contrast, necrosis is described as an uncontrolled and “accidental” process caused by exposure to overwhelming microenvironmental conditions such as extreme hyperthermia, hypothermia or radiation [6]. Necrotic cells exhibit swelling, plasma membrane rupture and increased permeability, as well as a loss of cellular contents and maintenance of DNA contents [7]. Necrosis is a strongly inflammatory process with little phagocytic involvement. ...
Fundamental properties of unperturbed haematopoiesis from stem cells in vivo
1
2015
... Cell death is one of the most fundamental physiological processes used by multicellular organisms to maintain homeostasis, as exemplified by tissue sculpting during embryologic development and the elimination of senescent cells. Cell death is also a characteristic of abnormal pathological responses to harmful stimuli, such as physical damage and pathogens [1]. Traditionally, the literature concerning cell death has been based on the morphology of dying cells and is dominated by apoptosis and necrosis [2]. Apoptosis, a process driven by the caspase family of enzymes, is the most studied and best characterized form of programmed cell death [3]. Morphological changes observed in apoptotic cells include DNA fragmentation, decreased cell volume and cytoplasmic and membrane vesicle formation [4]. Ultimately, apoptotic cells are cleared rapidly by adjacent phagocytes without inducing an inflammatory response. Because of these characteristics, apoptosis is considered to be “programmed” and “regulated” and is beneficial for normal embryonic development, tissue homeostasis and immune responses [5]. In contrast, necrosis is described as an uncontrolled and “accidental” process caused by exposure to overwhelming microenvironmental conditions such as extreme hyperthermia, hypothermia or radiation [6]. Necrotic cells exhibit swelling, plasma membrane rupture and increased permeability, as well as a loss of cellular contents and maintenance of DNA contents [7]. Necrosis is a strongly inflammatory process with little phagocytic involvement. ...
Live to die another way: modes of programmed cell death and the signals emanating from dying cells
1
2015
... Cell death is one of the most fundamental physiological processes used by multicellular organisms to maintain homeostasis, as exemplified by tissue sculpting during embryologic development and the elimination of senescent cells. Cell death is also a characteristic of abnormal pathological responses to harmful stimuli, such as physical damage and pathogens [1]. Traditionally, the literature concerning cell death has been based on the morphology of dying cells and is dominated by apoptosis and necrosis [2]. Apoptosis, a process driven by the caspase family of enzymes, is the most studied and best characterized form of programmed cell death [3]. Morphological changes observed in apoptotic cells include DNA fragmentation, decreased cell volume and cytoplasmic and membrane vesicle formation [4]. Ultimately, apoptotic cells are cleared rapidly by adjacent phagocytes without inducing an inflammatory response. Because of these characteristics, apoptosis is considered to be “programmed” and “regulated” and is beneficial for normal embryonic development, tissue homeostasis and immune responses [5]. In contrast, necrosis is described as an uncontrolled and “accidental” process caused by exposure to overwhelming microenvironmental conditions such as extreme hyperthermia, hypothermia or radiation [6]. Necrotic cells exhibit swelling, plasma membrane rupture and increased permeability, as well as a loss of cellular contents and maintenance of DNA contents [7]. Necrosis is a strongly inflammatory process with little phagocytic involvement. ...
Control of infection by pyroptosis and autophagy: role of TLR and NLR
1
2010
... Cell death is one of the most fundamental physiological processes used by multicellular organisms to maintain homeostasis, as exemplified by tissue sculpting during embryologic development and the elimination of senescent cells. Cell death is also a characteristic of abnormal pathological responses to harmful stimuli, such as physical damage and pathogens [1]. Traditionally, the literature concerning cell death has been based on the morphology of dying cells and is dominated by apoptosis and necrosis [2]. Apoptosis, a process driven by the caspase family of enzymes, is the most studied and best characterized form of programmed cell death [3]. Morphological changes observed in apoptotic cells include DNA fragmentation, decreased cell volume and cytoplasmic and membrane vesicle formation [4]. Ultimately, apoptotic cells are cleared rapidly by adjacent phagocytes without inducing an inflammatory response. Because of these characteristics, apoptosis is considered to be “programmed” and “regulated” and is beneficial for normal embryonic development, tissue homeostasis and immune responses [5]. In contrast, necrosis is described as an uncontrolled and “accidental” process caused by exposure to overwhelming microenvironmental conditions such as extreme hyperthermia, hypothermia or radiation [6]. Necrotic cells exhibit swelling, plasma membrane rupture and increased permeability, as well as a loss of cellular contents and maintenance of DNA contents [7]. Necrosis is a strongly inflammatory process with little phagocytic involvement. ...
Essential versus accessory aspects of cell death: recommendations of the NCCD 2015
1
2015
... Cell death is one of the most fundamental physiological processes used by multicellular organisms to maintain homeostasis, as exemplified by tissue sculpting during embryologic development and the elimination of senescent cells. Cell death is also a characteristic of abnormal pathological responses to harmful stimuli, such as physical damage and pathogens [1]. Traditionally, the literature concerning cell death has been based on the morphology of dying cells and is dominated by apoptosis and necrosis [2]. Apoptosis, a process driven by the caspase family of enzymes, is the most studied and best characterized form of programmed cell death [3]. Morphological changes observed in apoptotic cells include DNA fragmentation, decreased cell volume and cytoplasmic and membrane vesicle formation [4]. Ultimately, apoptotic cells are cleared rapidly by adjacent phagocytes without inducing an inflammatory response. Because of these characteristics, apoptosis is considered to be “programmed” and “regulated” and is beneficial for normal embryonic development, tissue homeostasis and immune responses [5]. In contrast, necrosis is described as an uncontrolled and “accidental” process caused by exposure to overwhelming microenvironmental conditions such as extreme hyperthermia, hypothermia or radiation [6]. Necrotic cells exhibit swelling, plasma membrane rupture and increased permeability, as well as a loss of cellular contents and maintenance of DNA contents [7]. Necrosis is a strongly inflammatory process with little phagocytic involvement. ...
Apoptosis, oncosis, and necrosis. An overview of cell death
1
1995
... Cell death is one of the most fundamental physiological processes used by multicellular organisms to maintain homeostasis, as exemplified by tissue sculpting during embryologic development and the elimination of senescent cells. Cell death is also a characteristic of abnormal pathological responses to harmful stimuli, such as physical damage and pathogens [1]. Traditionally, the literature concerning cell death has been based on the morphology of dying cells and is dominated by apoptosis and necrosis [2]. Apoptosis, a process driven by the caspase family of enzymes, is the most studied and best characterized form of programmed cell death [3]. Morphological changes observed in apoptotic cells include DNA fragmentation, decreased cell volume and cytoplasmic and membrane vesicle formation [4]. Ultimately, apoptotic cells are cleared rapidly by adjacent phagocytes without inducing an inflammatory response. Because of these characteristics, apoptosis is considered to be “programmed” and “regulated” and is beneficial for normal embryonic development, tissue homeostasis and immune responses [5]. In contrast, necrosis is described as an uncontrolled and “accidental” process caused by exposure to overwhelming microenvironmental conditions such as extreme hyperthermia, hypothermia or radiation [6]. Necrotic cells exhibit swelling, plasma membrane rupture and increased permeability, as well as a loss of cellular contents and maintenance of DNA contents [7]. Necrosis is a strongly inflammatory process with little phagocytic involvement. ...
MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death
1
2017
... Cell death is one of the most fundamental physiological processes used by multicellular organisms to maintain homeostasis, as exemplified by tissue sculpting during embryologic development and the elimination of senescent cells. Cell death is also a characteristic of abnormal pathological responses to harmful stimuli, such as physical damage and pathogens [1]. Traditionally, the literature concerning cell death has been based on the morphology of dying cells and is dominated by apoptosis and necrosis [2]. Apoptosis, a process driven by the caspase family of enzymes, is the most studied and best characterized form of programmed cell death [3]. Morphological changes observed in apoptotic cells include DNA fragmentation, decreased cell volume and cytoplasmic and membrane vesicle formation [4]. Ultimately, apoptotic cells are cleared rapidly by adjacent phagocytes without inducing an inflammatory response. Because of these characteristics, apoptosis is considered to be “programmed” and “regulated” and is beneficial for normal embryonic development, tissue homeostasis and immune responses [5]. In contrast, necrosis is described as an uncontrolled and “accidental” process caused by exposure to overwhelming microenvironmental conditions such as extreme hyperthermia, hypothermia or radiation [6]. Necrotic cells exhibit swelling, plasma membrane rupture and increased permeability, as well as a loss of cellular contents and maintenance of DNA contents [7]. Necrosis is a strongly inflammatory process with little phagocytic involvement. ...
Cell-Cycle Regulators and Cell Death in Immunity
1
2015
... Recent studies have revealed several other types of cell death, including autophagy [8], necroptosis [9], pyroptosis [10], ferroptosis [11], mitochondrial permeability transition (MPT)-dependent necrosis [12] and parthanatos [13]. These discoveries have led to the concept that multiple forms of regulated cell death can encompass morphological and biochemical features similar to both apoptosis and necrosis [14]. Pyroptosis is a type of proinflammatory programmed cell death that is triggered by inflammasomes. This process induces cell rupture and the release of cell contents, similar to necrosis, but pyroptosis involves caspase-driven programmed cell death, which is consistent with apoptosis [15]. ...
Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule
1
2000
... Recent studies have revealed several other types of cell death, including autophagy [8], necroptosis [9], pyroptosis [10], ferroptosis [11], mitochondrial permeability transition (MPT)-dependent necrosis [12] and parthanatos [13]. These discoveries have led to the concept that multiple forms of regulated cell death can encompass morphological and biochemical features similar to both apoptosis and necrosis [14]. Pyroptosis is a type of proinflammatory programmed cell death that is triggered by inflammasomes. This process induces cell rupture and the release of cell contents, similar to necrosis, but pyroptosis involves caspase-driven programmed cell death, which is consistent with apoptosis [15]. ...
Pyroptosis and its relationship to atherosclerosis
1
2018
... Recent studies have revealed several other types of cell death, including autophagy [8], necroptosis [9], pyroptosis [10], ferroptosis [11], mitochondrial permeability transition (MPT)-dependent necrosis [12] and parthanatos [13]. These discoveries have led to the concept that multiple forms of regulated cell death can encompass morphological and biochemical features similar to both apoptosis and necrosis [14]. Pyroptosis is a type of proinflammatory programmed cell death that is triggered by inflammasomes. This process induces cell rupture and the release of cell contents, similar to necrosis, but pyroptosis involves caspase-driven programmed cell death, which is consistent with apoptosis [15]. ...
Ferroptosis: an iron-dependent form of nonapoptotic cell death
1
2012
... Recent studies have revealed several other types of cell death, including autophagy [8], necroptosis [9], pyroptosis [10], ferroptosis [11], mitochondrial permeability transition (MPT)-dependent necrosis [12] and parthanatos [13]. These discoveries have led to the concept that multiple forms of regulated cell death can encompass morphological and biochemical features similar to both apoptosis and necrosis [14]. Pyroptosis is a type of proinflammatory programmed cell death that is triggered by inflammasomes. This process induces cell rupture and the release of cell contents, similar to necrosis, but pyroptosis involves caspase-driven programmed cell death, which is consistent with apoptosis [15]. ...
Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death
1
2005
... Recent studies have revealed several other types of cell death, including autophagy [8], necroptosis [9], pyroptosis [10], ferroptosis [11], mitochondrial permeability transition (MPT)-dependent necrosis [12] and parthanatos [13]. These discoveries have led to the concept that multiple forms of regulated cell death can encompass morphological and biochemical features similar to both apoptosis and necrosis [14]. Pyroptosis is a type of proinflammatory programmed cell death that is triggered by inflammasomes. This process induces cell rupture and the release of cell contents, similar to necrosis, but pyroptosis involves caspase-driven programmed cell death, which is consistent with apoptosis [15]. ...
Mitochondrial and nuclear cross talk in cell death: parthanatos
1
2008
... Recent studies have revealed several other types of cell death, including autophagy [8], necroptosis [9], pyroptosis [10], ferroptosis [11], mitochondrial permeability transition (MPT)-dependent necrosis [12] and parthanatos [13]. These discoveries have led to the concept that multiple forms of regulated cell death can encompass morphological and biochemical features similar to both apoptosis and necrosis [14]. Pyroptosis is a type of proinflammatory programmed cell death that is triggered by inflammasomes. This process induces cell rupture and the release of cell contents, similar to necrosis, but pyroptosis involves caspase-driven programmed cell death, which is consistent with apoptosis [15]. ...
Pyroptosis: host cell death and inflammation
1
2009
... Recent studies have revealed several other types of cell death, including autophagy [8], necroptosis [9], pyroptosis [10], ferroptosis [11], mitochondrial permeability transition (MPT)-dependent necrosis [12] and parthanatos [13]. These discoveries have led to the concept that multiple forms of regulated cell death can encompass morphological and biochemical features similar to both apoptosis and necrosis [14]. Pyroptosis is a type of proinflammatory programmed cell death that is triggered by inflammasomes. This process induces cell rupture and the release of cell contents, similar to necrosis, but pyroptosis involves caspase-driven programmed cell death, which is consistent with apoptosis [15]. ...
Pro-inflammatory programmed cell death
1
2001
... Recent studies have revealed several other types of cell death, including autophagy [8], necroptosis [9], pyroptosis [10], ferroptosis [11], mitochondrial permeability transition (MPT)-dependent necrosis [12] and parthanatos [13]. These discoveries have led to the concept that multiple forms of regulated cell death can encompass morphological and biochemical features similar to both apoptosis and necrosis [14]. Pyroptosis is a type of proinflammatory programmed cell death that is triggered by inflammasomes. This process induces cell rupture and the release of cell contents, similar to necrosis, but pyroptosis involves caspase-driven programmed cell death, which is consistent with apoptosis [15]. ...
Osteoarthritis: toward a comprehensive understanding of pathological mechanism
1
2017
... Osteoarthritis (OA) is the most common chronic degenerative disease worldwide. Although OA is highly prevalent [16], it mainly affects elderly people. This disease can involve nearly any joint in the human body, and the most common symptoms include joint pain and disordered articular functions [17]. The pathological changes associated with OA affect all tissues in the joint and include cartilage degeneration, subchondral sclerosis, variable degrees of synovial inflammation, osteophyte formation and hypertrophy of the whole joint capsule [18]. ...
Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis
1
2015
... Osteoarthritis (OA) is the most common chronic degenerative disease worldwide. Although OA is highly prevalent [16], it mainly affects elderly people. This disease can involve nearly any joint in the human body, and the most common symptoms include joint pain and disordered articular functions [17]. The pathological changes associated with OA affect all tissues in the joint and include cartilage degeneration, subchondral sclerosis, variable degrees of synovial inflammation, osteophyte formation and hypertrophy of the whole joint capsule [18]. ...
Current research on pharmacologic and regenerative therapies for osteoarthritis
1
2016
... Osteoarthritis (OA) is the most common chronic degenerative disease worldwide. Although OA is highly prevalent [16], it mainly affects elderly people. This disease can involve nearly any joint in the human body, and the most common symptoms include joint pain and disordered articular functions [17]. The pathological changes associated with OA affect all tissues in the joint and include cartilage degeneration, subchondral sclerosis, variable degrees of synovial inflammation, osteophyte formation and hypertrophy of the whole joint capsule [18]. ...
NLRP3 as a potentially novel biomarker for the management of osteoarthritis
2
2018
... Inflammasomes such as NLRP3 (NLR family, pyrin domain containing 3; NLR refers to “nucleotide-binding domain, leucine-rich repeat”), which are induced by nuclear factor kappa B (NF-κB) signaling and can convert interleukin-1β (IL-1β) and IL-18 into mature proinflammatory cytokines, are considered a factor in low-grade inflammatory pathology [19]. Among these inflammasomes, which comprise NLRs (NLRP1, NLRP3, and NLRC4), absent in melanoma 2 (AIM2), IFN-inducible protein 16 (IFI16) and pyrin, the NLRP3 inflammasome is the best characterized. This complex comprises NLRP3, apoptosis-related speck-like protein containing (ASC) a caspase recruitment domain (CARD) and procaspase-1 [20], suggesting that pyroptosis may be closely involved in the pathological changes associated with OA. Furthermore, the frequency of OA pain was indicated to be mediated by aberrantly overexpressed cytokines and to be associated strongly with inflammatory severity, especially in soft tissues such as the synovium, which could lead to hypersensitivity with exaggerated pain (hyperalgesia) in response to noxious stimuli or innocuous stimuli that are perceived as painful (allodynia) [21]. This indicates that pyroptosis may contribute to OA pain through releasing related cytokines [22]. In this review, we aimed to identify the possible mechanism by which pyroptosis contributes to OA progression and pain. Because no efficient treatment option is currently available for OA, an understanding of pyroptosis and related signaling factors might be useful in clinical decision-making. ...
... Existing evidence suggests that inflammasomes such as NLRP3 can be activated by fatty acids and high glucose levels, leading to chronic intestinal inflammation [59] and colorectal cancer [60]. These findings indicate that pyroptosis plays a significant role in digestive tract inflammation as well as in obesity-associated carcinogenesis. Pyroptosis, a highly inflammatory mode of cell death, can also trigger autoimmune diseases such as systemic lupus erythematosus (SLE) [61]. LPS from gram-negative bacterial infection induce pyroptosis by activating caspase-4/5/11, contributing to infectious diseases such as sepsis [25]. Pyroptosis also plays a role in inflammatory joint diseases such as rheumatoid arthritis (RA) [62] and OA [19]. ...
NLRP3 inflammasome and its inhibitors: a review
1
2015
... Inflammasomes such as NLRP3 (NLR family, pyrin domain containing 3; NLR refers to “nucleotide-binding domain, leucine-rich repeat”), which are induced by nuclear factor kappa B (NF-κB) signaling and can convert interleukin-1β (IL-1β) and IL-18 into mature proinflammatory cytokines, are considered a factor in low-grade inflammatory pathology [19]. Among these inflammasomes, which comprise NLRs (NLRP1, NLRP3, and NLRC4), absent in melanoma 2 (AIM2), IFN-inducible protein 16 (IFI16) and pyrin, the NLRP3 inflammasome is the best characterized. This complex comprises NLRP3, apoptosis-related speck-like protein containing (ASC) a caspase recruitment domain (CARD) and procaspase-1 [20], suggesting that pyroptosis may be closely involved in the pathological changes associated with OA. Furthermore, the frequency of OA pain was indicated to be mediated by aberrantly overexpressed cytokines and to be associated strongly with inflammatory severity, especially in soft tissues such as the synovium, which could lead to hypersensitivity with exaggerated pain (hyperalgesia) in response to noxious stimuli or innocuous stimuli that are perceived as painful (allodynia) [21]. This indicates that pyroptosis may contribute to OA pain through releasing related cytokines [22]. In this review, we aimed to identify the possible mechanism by which pyroptosis contributes to OA progression and pain. Because no efficient treatment option is currently available for OA, an understanding of pyroptosis and related signaling factors might be useful in clinical decision-making. ...
Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain
2
2019
... Inflammasomes such as NLRP3 (NLR family, pyrin domain containing 3; NLR refers to “nucleotide-binding domain, leucine-rich repeat”), which are induced by nuclear factor kappa B (NF-κB) signaling and can convert interleukin-1β (IL-1β) and IL-18 into mature proinflammatory cytokines, are considered a factor in low-grade inflammatory pathology [19]. Among these inflammasomes, which comprise NLRs (NLRP1, NLRP3, and NLRC4), absent in melanoma 2 (AIM2), IFN-inducible protein 16 (IFI16) and pyrin, the NLRP3 inflammasome is the best characterized. This complex comprises NLRP3, apoptosis-related speck-like protein containing (ASC) a caspase recruitment domain (CARD) and procaspase-1 [20], suggesting that pyroptosis may be closely involved in the pathological changes associated with OA. Furthermore, the frequency of OA pain was indicated to be mediated by aberrantly overexpressed cytokines and to be associated strongly with inflammatory severity, especially in soft tissues such as the synovium, which could lead to hypersensitivity with exaggerated pain (hyperalgesia) in response to noxious stimuli or innocuous stimuli that are perceived as painful (allodynia) [21]. This indicates that pyroptosis may contribute to OA pain through releasing related cytokines [22]. In this review, we aimed to identify the possible mechanism by which pyroptosis contributes to OA progression and pain. Because no efficient treatment option is currently available for OA, an understanding of pyroptosis and related signaling factors might be useful in clinical decision-making. ...
... Because its exact pathogenesis has not been fully revealed, OA is characterized as a whole-joint disease associated with pathological changes in all involved tissues. The pathology of OA includes the progressive loss and destruction of cartilage, sclerosis of the subchondral bone, formation of osteophytes, variable degrees of synovial inflammation, degeneration of the ligaments and meniscus and hypertrophy of the whole joint capsule[73, 74]. Initially, the risk factors that cause joint instability, such as ligament injury and excessive body weight, induce the excessive secretion of transforming growth factor β1 (TGF-β1) in the subchondral bone. An excess of this cytokine leads to the uncoupling of bone formation and resorption, which is accompanied by angiogenesis, sensory innervation [21], bone cavity formation and sclerosis [75]. Articular cartilage degeneration is the primary concern in OA [76]. Normally, in an environment with low oxygen, chondrocytes exhibit a low-grade turnover rate to maintain an anabolic-catabolic balance in the cartilage [77]. In OA, however, the expression of genes that encode proteins associated with inflammatory and catabolic responses is upregulated primarily via signal transduction pathways involving NF-κB, mitogen-activated protein kinase and other factors that are activated by inflammation. This transcriptional activity increases the production of primary inflammatory cytokines, such as IL-1β and tumor necrosis factor α (TNF-α), which play critical roles in OA pathology. IL-1β and TNF-α stimulate chondrocytes to release cartilage-degrading enzymes such as metalloproteinases 1, 3 and 13 (MMP1, MMP3 and MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 [78, 79]. These enzymes digest aggrecans and type II collagen to degrade the cartilage matrix. IL-1β and TNF-α also contribute to inflammatory chondrocyte death [80]. Together, these processes lead to a gradual loss of cartilage and the formation of osteochondral fissures, which allow the formation of new endochondral bone accompanied by angiogenesis and sensory innervation [81]. These processes may be closely associated with OA pain. During the late stage of OA, patients lose joint function and present with an immensely narrowed joint space containing little remaining cartilage, especially in the knee and hip joints. ...
Mechanisms of chronic pain in osteoarthritis
1
2012
... Inflammasomes such as NLRP3 (NLR family, pyrin domain containing 3; NLR refers to “nucleotide-binding domain, leucine-rich repeat”), which are induced by nuclear factor kappa B (NF-κB) signaling and can convert interleukin-1β (IL-1β) and IL-18 into mature proinflammatory cytokines, are considered a factor in low-grade inflammatory pathology [19]. Among these inflammasomes, which comprise NLRs (NLRP1, NLRP3, and NLRC4), absent in melanoma 2 (AIM2), IFN-inducible protein 16 (IFI16) and pyrin, the NLRP3 inflammasome is the best characterized. This complex comprises NLRP3, apoptosis-related speck-like protein containing (ASC) a caspase recruitment domain (CARD) and procaspase-1 [20], suggesting that pyroptosis may be closely involved in the pathological changes associated with OA. Furthermore, the frequency of OA pain was indicated to be mediated by aberrantly overexpressed cytokines and to be associated strongly with inflammatory severity, especially in soft tissues such as the synovium, which could lead to hypersensitivity with exaggerated pain (hyperalgesia) in response to noxious stimuli or innocuous stimuli that are perceived as painful (allodynia) [21]. This indicates that pyroptosis may contribute to OA pain through releasing related cytokines [22]. In this review, we aimed to identify the possible mechanism by which pyroptosis contributes to OA progression and pain. Because no efficient treatment option is currently available for OA, an understanding of pyroptosis and related signaling factors might be useful in clinical decision-making. ...
Salmonella-induced cell death: apoptosis, necrosis or programmed cell death?
1
2001
... Classic apoptosis is a caspase-dependent process of programmed cell death. An alternative form of cell death involving caspase-dependent programmed cell death combined with proinflammatory changes has also been identified [23]. This process was termed “pyroptosis” [24] after the identification of macrophages infected with Salmonella or Shigella spp. The term for this type of proinflammatory programmed cell death was derived from the Greek roots pyro, meaning “fire” or “fever,” and ptosis, which means “falling” [25]. In contrast to other forms of programmed cell death, pyroptosis is closely related to inflammation. Morphologically, pyroptosis involves the cleavage of procaspase-1 to yield active caspase-1. This activated enzyme forms pores in the plasma membrane, which enables the equalization of the ionic gradient between the intracellular and extracellular environments. Consequently, water flows into the cell, causing swelling and lysis [26]. Along with the increase in cell size and formation of a balloon-like surrounding vesicle, the nucleus condenses and becomes rounded [27, 28]. Although DNA fragmentation consistent with apoptosis is observed, nuclear integrity is maintained in pyroptosis [29, 30]. Therefore, the morphological changes associated with pyroptosis are distinct from those associated with apoptosis, and the former process can be described as having a combination of apoptotic and necrotic features. ...
Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells
1
2005
... Classic apoptosis is a caspase-dependent process of programmed cell death. An alternative form of cell death involving caspase-dependent programmed cell death combined with proinflammatory changes has also been identified [23]. This process was termed “pyroptosis” [24] after the identification of macrophages infected with Salmonella or Shigella spp. The term for this type of proinflammatory programmed cell death was derived from the Greek roots pyro, meaning “fire” or “fever,” and ptosis, which means “falling” [25]. In contrast to other forms of programmed cell death, pyroptosis is closely related to inflammation. Morphologically, pyroptosis involves the cleavage of procaspase-1 to yield active caspase-1. This activated enzyme forms pores in the plasma membrane, which enables the equalization of the ionic gradient between the intracellular and extracellular environments. Consequently, water flows into the cell, causing swelling and lysis [26]. Along with the increase in cell size and formation of a balloon-like surrounding vesicle, the nucleus condenses and becomes rounded [27, 28]. Although DNA fragmentation consistent with apoptosis is observed, nuclear integrity is maintained in pyroptosis [29, 30]. Therefore, the morphological changes associated with pyroptosis are distinct from those associated with apoptosis, and the former process can be described as having a combination of apoptotic and necrotic features. ...
Recent Advances in the Molecular Mechanisms Underlying Pyroptosis in Sepsis
2
2018
... Classic apoptosis is a caspase-dependent process of programmed cell death. An alternative form of cell death involving caspase-dependent programmed cell death combined with proinflammatory changes has also been identified [23]. This process was termed “pyroptosis” [24] after the identification of macrophages infected with Salmonella or Shigella spp. The term for this type of proinflammatory programmed cell death was derived from the Greek roots pyro, meaning “fire” or “fever,” and ptosis, which means “falling” [25]. In contrast to other forms of programmed cell death, pyroptosis is closely related to inflammation. Morphologically, pyroptosis involves the cleavage of procaspase-1 to yield active caspase-1. This activated enzyme forms pores in the plasma membrane, which enables the equalization of the ionic gradient between the intracellular and extracellular environments. Consequently, water flows into the cell, causing swelling and lysis [26]. Along with the increase in cell size and formation of a balloon-like surrounding vesicle, the nucleus condenses and becomes rounded [27, 28]. Although DNA fragmentation consistent with apoptosis is observed, nuclear integrity is maintained in pyroptosis [29, 30]. Therefore, the morphological changes associated with pyroptosis are distinct from those associated with apoptosis, and the former process can be described as having a combination of apoptotic and necrotic features. ...
... Existing evidence suggests that inflammasomes such as NLRP3 can be activated by fatty acids and high glucose levels, leading to chronic intestinal inflammation [59] and colorectal cancer [60]. These findings indicate that pyroptosis plays a significant role in digestive tract inflammation as well as in obesity-associated carcinogenesis. Pyroptosis, a highly inflammatory mode of cell death, can also trigger autoimmune diseases such as systemic lupus erythematosus (SLE) [61]. LPS from gram-negative bacterial infection induce pyroptosis by activating caspase-4/5/11, contributing to infectious diseases such as sepsis [25]. Pyroptosis also plays a role in inflammatory joint diseases such as rheumatoid arthritis (RA) [62] and OA [19]. ...
The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1
1
1999
... Classic apoptosis is a caspase-dependent process of programmed cell death. An alternative form of cell death involving caspase-dependent programmed cell death combined with proinflammatory changes has also been identified [23]. This process was termed “pyroptosis” [24] after the identification of macrophages infected with Salmonella or Shigella spp. The term for this type of proinflammatory programmed cell death was derived from the Greek roots pyro, meaning “fire” or “fever,” and ptosis, which means “falling” [25]. In contrast to other forms of programmed cell death, pyroptosis is closely related to inflammation. Morphologically, pyroptosis involves the cleavage of procaspase-1 to yield active caspase-1. This activated enzyme forms pores in the plasma membrane, which enables the equalization of the ionic gradient between the intracellular and extracellular environments. Consequently, water flows into the cell, causing swelling and lysis [26]. Along with the increase in cell size and formation of a balloon-like surrounding vesicle, the nucleus condenses and becomes rounded [27, 28]. Although DNA fragmentation consistent with apoptosis is observed, nuclear integrity is maintained in pyroptosis [29, 30]. Therefore, the morphological changes associated with pyroptosis are distinct from those associated with apoptosis, and the former process can be described as having a combination of apoptotic and necrotic features. ...
The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation
1
2007
... Classic apoptosis is a caspase-dependent process of programmed cell death. An alternative form of cell death involving caspase-dependent programmed cell death combined with proinflammatory changes has also been identified [23]. This process was termed “pyroptosis” [24] after the identification of macrophages infected with Salmonella or Shigella spp. The term for this type of proinflammatory programmed cell death was derived from the Greek roots pyro, meaning “fire” or “fever,” and ptosis, which means “falling” [25]. In contrast to other forms of programmed cell death, pyroptosis is closely related to inflammation. Morphologically, pyroptosis involves the cleavage of procaspase-1 to yield active caspase-1. This activated enzyme forms pores in the plasma membrane, which enables the equalization of the ionic gradient between the intracellular and extracellular environments. Consequently, water flows into the cell, causing swelling and lysis [26]. Along with the increase in cell size and formation of a balloon-like surrounding vesicle, the nucleus condenses and becomes rounded [27, 28]. Although DNA fragmentation consistent with apoptosis is observed, nuclear integrity is maintained in pyroptosis [29, 30]. Therefore, the morphological changes associated with pyroptosis are distinct from those associated with apoptosis, and the former process can be described as having a combination of apoptotic and necrotic features. ...
Salmonella-induced caspase-2 activation in macrophages: a novel mechanism in pathogen-mediated apoptosis
1
2000
... Classic apoptosis is a caspase-dependent process of programmed cell death. An alternative form of cell death involving caspase-dependent programmed cell death combined with proinflammatory changes has also been identified [23]. This process was termed “pyroptosis” [24] after the identification of macrophages infected with Salmonella or Shigella spp. The term for this type of proinflammatory programmed cell death was derived from the Greek roots pyro, meaning “fire” or “fever,” and ptosis, which means “falling” [25]. In contrast to other forms of programmed cell death, pyroptosis is closely related to inflammation. Morphologically, pyroptosis involves the cleavage of procaspase-1 to yield active caspase-1. This activated enzyme forms pores in the plasma membrane, which enables the equalization of the ionic gradient between the intracellular and extracellular environments. Consequently, water flows into the cell, causing swelling and lysis [26]. Along with the increase in cell size and formation of a balloon-like surrounding vesicle, the nucleus condenses and becomes rounded [27, 28]. Although DNA fragmentation consistent with apoptosis is observed, nuclear integrity is maintained in pyroptosis [29, 30]. Therefore, the morphological changes associated with pyroptosis are distinct from those associated with apoptosis, and the former process can be described as having a combination of apoptotic and necrotic features. ...
Salmonella-induced cell death is not required for enteritis in calves
1
2001
... Classic apoptosis is a caspase-dependent process of programmed cell death. An alternative form of cell death involving caspase-dependent programmed cell death combined with proinflammatory changes has also been identified [23]. This process was termed “pyroptosis” [24] after the identification of macrophages infected with Salmonella or Shigella spp. The term for this type of proinflammatory programmed cell death was derived from the Greek roots pyro, meaning “fire” or “fever,” and ptosis, which means “falling” [25]. In contrast to other forms of programmed cell death, pyroptosis is closely related to inflammation. Morphologically, pyroptosis involves the cleavage of procaspase-1 to yield active caspase-1. This activated enzyme forms pores in the plasma membrane, which enables the equalization of the ionic gradient between the intracellular and extracellular environments. Consequently, water flows into the cell, causing swelling and lysis [26]. Along with the increase in cell size and formation of a balloon-like surrounding vesicle, the nucleus condenses and becomes rounded [27, 28]. Although DNA fragmentation consistent with apoptosis is observed, nuclear integrity is maintained in pyroptosis [29, 30]. Therefore, the morphological changes associated with pyroptosis are distinct from those associated with apoptosis, and the former process can be described as having a combination of apoptotic and necrotic features. ...
Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis
1
2005
... Classic apoptosis is a caspase-dependent process of programmed cell death. An alternative form of cell death involving caspase-dependent programmed cell death combined with proinflammatory changes has also been identified [23]. This process was termed “pyroptosis” [24] after the identification of macrophages infected with Salmonella or Shigella spp. The term for this type of proinflammatory programmed cell death was derived from the Greek roots pyro, meaning “fire” or “fever,” and ptosis, which means “falling” [25]. In contrast to other forms of programmed cell death, pyroptosis is closely related to inflammation. Morphologically, pyroptosis involves the cleavage of procaspase-1 to yield active caspase-1. This activated enzyme forms pores in the plasma membrane, which enables the equalization of the ionic gradient between the intracellular and extracellular environments. Consequently, water flows into the cell, causing swelling and lysis [26]. Along with the increase in cell size and formation of a balloon-like surrounding vesicle, the nucleus condenses and becomes rounded [27, 28]. Although DNA fragmentation consistent with apoptosis is observed, nuclear integrity is maintained in pyroptosis [29, 30]. Therefore, the morphological changes associated with pyroptosis are distinct from those associated with apoptosis, and the former process can be described as having a combination of apoptotic and necrotic features. ...
The inflammasomes
2
2010
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
... Aging is also a significant risk factor for multiple chronic diseases, as elderly people are more vulnerable due to their decreased immune and metabolic activities. A highly critical event occurs with aging, which starts with DAMP accumulation and activation of the subsequent NLRP3 inflammasome, which is stimulated by endogenous byproducts [103] that are then recognized by PRRs in macrophages to trigger chronic, low-grade inflammation [31]. In turn, chronic inflammation plays an important role in the progression of metabolic disorders, such as obesity, gouty arthritis and atherosclerosis [104]. ...
Inflammatory caspases and inflammasomes: master switches of inflammation
1
2007
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta
1
2002
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing
1
2010
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
Mechanisms and functions of inflammasomes
1
2014
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
Pattern recognition receptors and inflammation
1
2010
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
... Noncanonical inflammasomes detect intracellular bacteria and bacterial lipopolysaccharide (LPS) molecules in infected cells [48] and consequently activate caspase-11 (in mice) or caspase-4/5 (in humans). Subsequently, caspase-11 directly triggers pyroptosis. Once activated, these caspases cleave the cellular protein GSDMD, which inserts into the cell membrane lipid bilayer via its N-terminal domain. This process enables the formation of pores that induce osmotic cell lysis [49, 50]. A previous study proved that caspase-11 also indirectly processes IL-1β via the caspase-1 pathway [37], which suggests crosstalk between different caspases during the pyroptosis. ...
The NLRP3 inflammasome: molecular activation and regulation to therapeutics
1
2019
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression
1
2009
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
Cutting Edge: TRAF6 Mediates TLR/IL-1R Signaling-Induced Nontranscriptional Priming of the NLRP3 Inflammasome
2
2017
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
... ]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
Principles and properties of ion flow in P2X receptors
1
2014
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP
1
2012
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
Multiple Cathepsins Promote Pro-IL-1beta Synthesis and NLRP3-Mediated IL-1beta Activation
1
2015
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
Imidazoquinoxaline anticancer derivatives and imiquimod interact with tubulin: Characterization of molecular microtubule inhibiting mechanisms in correlation with cytotoxicity
1
2017
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation
1
2018
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells
1
2015
... Inflammasomes are multimeric protein complexes that assemble in the cytosol and act as platforms for caspase activation. These complexes play vital roles in the initiation of inflammation. Caspases are cysteine proteases that initiate or execute cellular programs. These proteases can induce inflammation or cell death, depending on their function [31] and are thus categorized as proinflammatory or proapoptotic. Proinflammatory caspases include caspases-1, -11 and -12 in mice and caspases-1, -4, and -5 in humans [32]. Of these, caspase-1, the most fully characterized, is regulated closely by the inflammasome to process cytokines [33]. Activated caspase-1 is required for proteolytic processing and for release of the cytokines IL-1β and IL-18 [34]. Caspases may be activated in response to various stimuli. Upon damage induced by endogenous stress or microbial infection, caspases release either danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that trigger pattern recognition receptors (PRRs) [35]. PRRs can be divided into two categories based on their subcellular localization. Transmembrane PRRs include Toll-like receptors (TLRs) and C-type lectins (CTLs), while intracellular PRRs include the RIG-I-like receptor (RLR), AIM2-like receptor (ALR) and nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins [36]. The pathways associated with pyroptosis are defined by the triggering of different caspases. The canonical inflammasome pathway is triggered by activated caspase-1, while the noncanonical inflammasome pathway is triggered by activated caspase-11 (in mice) or caspase-4/5 (in humans) [37]. In the canonical inflammasome pathway, assembly of the NLRP3 inflammasome in response to PAMPs or DAMPs can lead to caspase-1-dependent release of proinflammatory cytokines and gasdermin D (GSDMD)-mediated pyroptotic cell death. With few exceptions, NLRP3 inflammasome activation is considered to have two steps: priming and activation [38]. The priming step activates the inflammatory process in cells and upregulates the expression of inflammasome components upon increased transcriptional activity of nuclear factor-κB (NF-κB) [39, 40], upregulating the expression of inflammasome components, including NLRP3, caspase-1 and pro-IL-1β. The following step is activation of NLRP3 by a variety of upstream stimuli induced by numerous PAMPs or DAMPs, including K+efflux[41], Ca2+ flux [42], lysosomal disruption [43], mitochondrial dysfunction [44], metabolic changes [45] and trans- Golgi disassembly [46]. NLRP3 then interacts with NIMA (never in mitosis gene a)-related kinase 7 (NEK7) and forms a complex with ASC and caspase-1 (the NLRP3 inflammasome). The complex cleaves GSDMD to form GSDMD-N pores into the membrane, which induces pyroptosis. The complex also enhances the processing of inactive IL-1β and IL-18 into mature inflammatory cytokines, which can in turn promote the transcriptional activities of NF-κB, activator protein 1 (AP1) and interferon regulatory factors (IRFs) [40, 47]. ...
Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock
1
2013
... Noncanonical inflammasomes detect intracellular bacteria and bacterial lipopolysaccharide (LPS) molecules in infected cells [48] and consequently activate caspase-11 (in mice) or caspase-4/5 (in humans). Subsequently, caspase-11 directly triggers pyroptosis. Once activated, these caspases cleave the cellular protein GSDMD, which inserts into the cell membrane lipid bilayer via its N-terminal domain. This process enables the formation of pores that induce osmotic cell lysis [49, 50]. A previous study proved that caspase-11 also indirectly processes IL-1β via the caspase-1 pathway [37], which suggests crosstalk between different caspases during the pyroptosis. ...
Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling
1
2015
... Noncanonical inflammasomes detect intracellular bacteria and bacterial lipopolysaccharide (LPS) molecules in infected cells [48] and consequently activate caspase-11 (in mice) or caspase-4/5 (in humans). Subsequently, caspase-11 directly triggers pyroptosis. Once activated, these caspases cleave the cellular protein GSDMD, which inserts into the cell membrane lipid bilayer via its N-terminal domain. This process enables the formation of pores that induce osmotic cell lysis [49, 50]. A previous study proved that caspase-11 also indirectly processes IL-1β via the caspase-1 pathway [37], which suggests crosstalk between different caspases during the pyroptosis. ...
Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death
1
2015
... Noncanonical inflammasomes detect intracellular bacteria and bacterial lipopolysaccharide (LPS) molecules in infected cells [48] and consequently activate caspase-11 (in mice) or caspase-4/5 (in humans). Subsequently, caspase-11 directly triggers pyroptosis. Once activated, these caspases cleave the cellular protein GSDMD, which inserts into the cell membrane lipid bilayer via its N-terminal domain. This process enables the formation of pores that induce osmotic cell lysis [49, 50]. A previous study proved that caspase-11 also indirectly processes IL-1β via the caspase-1 pathway [37], which suggests crosstalk between different caspases during the pyroptosis. ...
Role of pyroptosis in cardiovascular disease
1
2019
... Several studies have investigated the roles of pyroptosis in different diseases, particularly cardiovascular diseases[51]. Existing evidence has revealed significant increases in caspase-1 expression in vulnerable plaques and ruptured lesions associated with acute coronary events in atherosclerosis [52]. Additionally, monocyte/ macrophage pyroptosis may also induce an amplified inflammatory response and promote plaque rupture and thrombosis, which can induce acute cardiovascular events [53]. Moreover, vascular smooth muscle cell pyroptosis may suppress the healing of vascular injuries and reduce the stability of atherosclerotic plaques [54, 55]. Cardiomyocyte and cardiac fibroblast pyroptosis is also involved in diabetic cardiomyopathy [56], cardiac hypertrophy [57] and ischemic heart disease [58]. ...
Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association
1
2017
... Several studies have investigated the roles of pyroptosis in different diseases, particularly cardiovascular diseases[51]. Existing evidence has revealed significant increases in caspase-1 expression in vulnerable plaques and ruptured lesions associated with acute coronary events in atherosclerosis [52]. Additionally, monocyte/ macrophage pyroptosis may also induce an amplified inflammatory response and promote plaque rupture and thrombosis, which can induce acute cardiovascular events [53]. Moreover, vascular smooth muscle cell pyroptosis may suppress the healing of vascular injuries and reduce the stability of atherosclerotic plaques [54, 55]. Cardiomyocyte and cardiac fibroblast pyroptosis is also involved in diabetic cardiomyopathy [56], cardiac hypertrophy [57] and ischemic heart disease [58]. ...
Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease
1
2011
... Several studies have investigated the roles of pyroptosis in different diseases, particularly cardiovascular diseases[51]. Existing evidence has revealed significant increases in caspase-1 expression in vulnerable plaques and ruptured lesions associated with acute coronary events in atherosclerosis [52]. Additionally, monocyte/ macrophage pyroptosis may also induce an amplified inflammatory response and promote plaque rupture and thrombosis, which can induce acute cardiovascular events [53]. Moreover, vascular smooth muscle cell pyroptosis may suppress the healing of vascular injuries and reduce the stability of atherosclerotic plaques [54, 55]. Cardiomyocyte and cardiac fibroblast pyroptosis is also involved in diabetic cardiomyopathy [56], cardiac hypertrophy [57] and ischemic heart disease [58]. ...
Phenotypic switching of vascular smooth muscle cells in the ’normal region’ of aorta from atherosclerosis patients is regulated by miR-145
1
2016
... Several studies have investigated the roles of pyroptosis in different diseases, particularly cardiovascular diseases[51]. Existing evidence has revealed significant increases in caspase-1 expression in vulnerable plaques and ruptured lesions associated with acute coronary events in atherosclerosis [52]. Additionally, monocyte/ macrophage pyroptosis may also induce an amplified inflammatory response and promote plaque rupture and thrombosis, which can induce acute cardiovascular events [53]. Moreover, vascular smooth muscle cell pyroptosis may suppress the healing of vascular injuries and reduce the stability of atherosclerotic plaques [54, 55]. Cardiomyocyte and cardiac fibroblast pyroptosis is also involved in diabetic cardiomyopathy [56], cardiac hypertrophy [57] and ischemic heart disease [58]. ...
Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis
1
2015
... Several studies have investigated the roles of pyroptosis in different diseases, particularly cardiovascular diseases[51]. Existing evidence has revealed significant increases in caspase-1 expression in vulnerable plaques and ruptured lesions associated with acute coronary events in atherosclerosis [52]. Additionally, monocyte/ macrophage pyroptosis may also induce an amplified inflammatory response and promote plaque rupture and thrombosis, which can induce acute cardiovascular events [53]. Moreover, vascular smooth muscle cell pyroptosis may suppress the healing of vascular injuries and reduce the stability of atherosclerotic plaques [54, 55]. Cardiomyocyte and cardiac fibroblast pyroptosis is also involved in diabetic cardiomyopathy [56], cardiac hypertrophy [57] and ischemic heart disease [58]. ...
NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model
1
2014
... Several studies have investigated the roles of pyroptosis in different diseases, particularly cardiovascular diseases[51]. Existing evidence has revealed significant increases in caspase-1 expression in vulnerable plaques and ruptured lesions associated with acute coronary events in atherosclerosis [52]. Additionally, monocyte/ macrophage pyroptosis may also induce an amplified inflammatory response and promote plaque rupture and thrombosis, which can induce acute cardiovascular events [53]. Moreover, vascular smooth muscle cell pyroptosis may suppress the healing of vascular injuries and reduce the stability of atherosclerotic plaques [54, 55]. Cardiomyocyte and cardiac fibroblast pyroptosis is also involved in diabetic cardiomyopathy [56], cardiac hypertrophy [57] and ischemic heart disease [58]. ...
Antihypertensive Therapies and Left Ventricular Hypertrophy
1
2017
... Several studies have investigated the roles of pyroptosis in different diseases, particularly cardiovascular diseases[51]. Existing evidence has revealed significant increases in caspase-1 expression in vulnerable plaques and ruptured lesions associated with acute coronary events in atherosclerosis [52]. Additionally, monocyte/ macrophage pyroptosis may also induce an amplified inflammatory response and promote plaque rupture and thrombosis, which can induce acute cardiovascular events [53]. Moreover, vascular smooth muscle cell pyroptosis may suppress the healing of vascular injuries and reduce the stability of atherosclerotic plaques [54, 55]. Cardiomyocyte and cardiac fibroblast pyroptosis is also involved in diabetic cardiomyopathy [56], cardiac hypertrophy [57] and ischemic heart disease [58]. ...
Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury
1
2011
... Several studies have investigated the roles of pyroptosis in different diseases, particularly cardiovascular diseases[51]. Existing evidence has revealed significant increases in caspase-1 expression in vulnerable plaques and ruptured lesions associated with acute coronary events in atherosclerosis [52]. Additionally, monocyte/ macrophage pyroptosis may also induce an amplified inflammatory response and promote plaque rupture and thrombosis, which can induce acute cardiovascular events [53]. Moreover, vascular smooth muscle cell pyroptosis may suppress the healing of vascular injuries and reduce the stability of atherosclerotic plaques [54, 55]. Cardiomyocyte and cardiac fibroblast pyroptosis is also involved in diabetic cardiomyopathy [56], cardiac hypertrophy [57] and ischemic heart disease [58]. ...
Inhibiting Interleukin 36 Receptor Signaling Reduces Fibrosis in Mice With Chronic Intestinal Inflammation
2
2019
... Existing evidence suggests that inflammasomes such as NLRP3 can be activated by fatty acids and high glucose levels, leading to chronic intestinal inflammation [59] and colorectal cancer [60]. These findings indicate that pyroptosis plays a significant role in digestive tract inflammation as well as in obesity-associated carcinogenesis. Pyroptosis, a highly inflammatory mode of cell death, can also trigger autoimmune diseases such as systemic lupus erythematosus (SLE) [61]. LPS from gram-negative bacterial infection induce pyroptosis by activating caspase-4/5/11, contributing to infectious diseases such as sepsis [25]. Pyroptosis also plays a role in inflammatory joint diseases such as rheumatoid arthritis (RA) [62] and OA [19]. ...
... Microcrystals, including basic calcium phosphate (BCP), calcium pyrophosphate and uric acid [98], are considered “danger signals” and are thought to be the main DAMPs that activate inflammasomes. BCP crystals have been found in the knee and hip joints of OA patients [99], and these crystals elicit the release of IL-1β from macrophages by activating NLRP3 in a process that is dependent on K+ efflux, reactive oxygen species (ROS) and phagocytosis [100, 101]. This pyroptosis-related process drives the pathology of OA and induces cartilage degeneration. Previous reports[102] suggested that in synovial fibroblasts and articular chondrocytes, IL-36, an IL-1 cytokine subfamily member, stimulates the expression of inflammatory mediators and thus plays a critical role in inflammatory diseases [59]. ...
Necroptosis: A new way of dying?
1
2016
... Existing evidence suggests that inflammasomes such as NLRP3 can be activated by fatty acids and high glucose levels, leading to chronic intestinal inflammation [59] and colorectal cancer [60]. These findings indicate that pyroptosis plays a significant role in digestive tract inflammation as well as in obesity-associated carcinogenesis. Pyroptosis, a highly inflammatory mode of cell death, can also trigger autoimmune diseases such as systemic lupus erythematosus (SLE) [61]. LPS from gram-negative bacterial infection induce pyroptosis by activating caspase-4/5/11, contributing to infectious diseases such as sepsis [25]. Pyroptosis also plays a role in inflammatory joint diseases such as rheumatoid arthritis (RA) [62] and OA [19]. ...
The Role of Cell Death in the Pathogenesis of SLE: Is Pyroptosis the Missing Link?
1
2015
... Existing evidence suggests that inflammasomes such as NLRP3 can be activated by fatty acids and high glucose levels, leading to chronic intestinal inflammation [59] and colorectal cancer [60]. These findings indicate that pyroptosis plays a significant role in digestive tract inflammation as well as in obesity-associated carcinogenesis. Pyroptosis, a highly inflammatory mode of cell death, can also trigger autoimmune diseases such as systemic lupus erythematosus (SLE) [61]. LPS from gram-negative bacterial infection induce pyroptosis by activating caspase-4/5/11, contributing to infectious diseases such as sepsis [25]. Pyroptosis also plays a role in inflammatory joint diseases such as rheumatoid arthritis (RA) [62] and OA [19]. ...
Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment
2
2014
... Existing evidence suggests that inflammasomes such as NLRP3 can be activated by fatty acids and high glucose levels, leading to chronic intestinal inflammation [59] and colorectal cancer [60]. These findings indicate that pyroptosis plays a significant role in digestive tract inflammation as well as in obesity-associated carcinogenesis. Pyroptosis, a highly inflammatory mode of cell death, can also trigger autoimmune diseases such as systemic lupus erythematosus (SLE) [61]. LPS from gram-negative bacterial infection induce pyroptosis by activating caspase-4/5/11, contributing to infectious diseases such as sepsis [25]. Pyroptosis also plays a role in inflammatory joint diseases such as rheumatoid arthritis (RA) [62] and OA [19]. ...
... Chronic synovial inflammation is a known cause of cartilage degradation [87, 107]. In OA, IL-1β, which is involved in cartilage degradation, may be produced by synovial tissue rather than by chondrocytes [108]. Furthermore, the protein level of NLRP3 is more than five-fold higher in the synovial membranes of OA patients than in subjects with normal joints [109]. Genetic studies have proven a close association of inflammasomes with RA, which is a primary autoimmune disease of the synovium. These studies revealed the significantly upregulated expression of genes encoding NLRP3 inflammasome components, including NLRP3, ASC and caspase-1, in patients with RA [62, 110]. ...
Osteoarthritis
2
2016
... As noted previously, OA is the most common degenerative joint disorder and can affect nearly any small or large joint in the body [63]. OA is now considered to have complex etiology, and these multiple factors can lead to similar outcomes of joint destruction and clinical features [64]. In a study of OA patients older than 50 years of age in North America and Europe, approximately 60%, 33% and 5% reported involvement of joints of the hand, knee and hip, respectively [65]. An age older than 40 years is associated with a greater likelihood of developing OA. Moreover, OA is more frequent in women than in men at any age older than 50 years (i.e., postmenopausal women), and this sex difference mainly involves the hand (e.g., distal interphalangeal) and knee joints [66]. The risk factors for OA include factors related to individual susceptibility, as well as those affecting biomechanical joint stability [67]. Obesity may also be a risk factor for OA, as it increases stress on weight-bearing joints such as the hips and knees. Additionally, fat cells produce potentially harmful inflammatory proteins that may target the joints. The genetics underlying OA are complex but highly significant and may be related to changes in important molecular pathways. The genes encoding inflammatory and catabolism-related proteins are upregulated during OA, primarily through signal transduction involving nuclear factor-κB (NF-κB), mitogen- activated protein kinase, and other inflammation- and stress-induced pathways [63, 68]. Joint injuries, such as those involving the ligaments and meniscus, can increase the fragility of the joint and increase the risk of osteoarthritis. Even injuries that occurred many years ago can increase the risk of OA. Moreover, joint malformation or cartilage defects can increase the risk of OA [69-71]. The symptoms of OA include pain, transient morning stiffness and a grating sensation when walking. End-stage OA is characterized by instability and physical disability, which have tremendous negative effects on the quality of life [72]. The lack of efficient and curative treatment options for OA and the ever-evolving pathophysiological and risk factors present significant challenges. ...
... ]. Obesity may also be a risk factor for OA, as it increases stress on weight-bearing joints such as the hips and knees. Additionally, fat cells produce potentially harmful inflammatory proteins that may target the joints. The genetics underlying OA are complex but highly significant and may be related to changes in important molecular pathways. The genes encoding inflammatory and catabolism-related proteins are upregulated during OA, primarily through signal transduction involving nuclear factor-κB (NF-κB), mitogen- activated protein kinase, and other inflammation- and stress-induced pathways [63, 68]. Joint injuries, such as those involving the ligaments and meniscus, can increase the fragility of the joint and increase the risk of osteoarthritis. Even injuries that occurred many years ago can increase the risk of OA. Moreover, joint malformation or cartilage defects can increase the risk of OA [69-71]. The symptoms of OA include pain, transient morning stiffness and a grating sensation when walking. End-stage OA is characterized by instability and physical disability, which have tremendous negative effects on the quality of life [72]. The lack of efficient and curative treatment options for OA and the ever-evolving pathophysiological and risk factors present significant challenges. ...
Metabolic triggered inflammation in osteoarthritis
2
2015
... As noted previously, OA is the most common degenerative joint disorder and can affect nearly any small or large joint in the body [63]. OA is now considered to have complex etiology, and these multiple factors can lead to similar outcomes of joint destruction and clinical features [64]. In a study of OA patients older than 50 years of age in North America and Europe, approximately 60%, 33% and 5% reported involvement of joints of the hand, knee and hip, respectively [65]. An age older than 40 years is associated with a greater likelihood of developing OA. Moreover, OA is more frequent in women than in men at any age older than 50 years (i.e., postmenopausal women), and this sex difference mainly involves the hand (e.g., distal interphalangeal) and knee joints [66]. The risk factors for OA include factors related to individual susceptibility, as well as those affecting biomechanical joint stability [67]. Obesity may also be a risk factor for OA, as it increases stress on weight-bearing joints such as the hips and knees. Additionally, fat cells produce potentially harmful inflammatory proteins that may target the joints. The genetics underlying OA are complex but highly significant and may be related to changes in important molecular pathways. The genes encoding inflammatory and catabolism-related proteins are upregulated during OA, primarily through signal transduction involving nuclear factor-κB (NF-κB), mitogen- activated protein kinase, and other inflammation- and stress-induced pathways [63, 68]. Joint injuries, such as those involving the ligaments and meniscus, can increase the fragility of the joint and increase the risk of osteoarthritis. Even injuries that occurred many years ago can increase the risk of OA. Moreover, joint malformation or cartilage defects can increase the risk of OA [69-71]. The symptoms of OA include pain, transient morning stiffness and a grating sensation when walking. End-stage OA is characterized by instability and physical disability, which have tremendous negative effects on the quality of life [72]. The lack of efficient and curative treatment options for OA and the ever-evolving pathophysiological and risk factors present significant challenges. ...
... Metabolic disorders contribute to OA pathogenesis. Obesity (body mass index > 30 kg/m2) is a significant risk factor for the onset and progression of OA [91]. Waist circumference, which is an indicator of central adiposity, is associated with increased knee cartilage defects [92], reduced knee cartilage mass and increased bone marrow lesions [93]. Adipokines, one of the obesity-related metabolic factors, have been shown to induce cartilage degradation [64]. Leptin, an adipokine, has a detrimental effect on chondrocyte proliferation by increasing IL-1β and MMPs [94]. Additionally, individuals with obesity have low circulating 25-hydroxy-vitamin D (25-(OH)D) [95], which is positively associated with the development and worsening of knee OA, including cartilage loss and increased OA pain [96]. ...
The prevalence of chondrocalcinosis in the elderly and its association with knee osteoarthritis: the Framingham Study
1
1989
... As noted previously, OA is the most common degenerative joint disorder and can affect nearly any small or large joint in the body [63]. OA is now considered to have complex etiology, and these multiple factors can lead to similar outcomes of joint destruction and clinical features [64]. In a study of OA patients older than 50 years of age in North America and Europe, approximately 60%, 33% and 5% reported involvement of joints of the hand, knee and hip, respectively [65]. An age older than 40 years is associated with a greater likelihood of developing OA. Moreover, OA is more frequent in women than in men at any age older than 50 years (i.e., postmenopausal women), and this sex difference mainly involves the hand (e.g., distal interphalangeal) and knee joints [66]. The risk factors for OA include factors related to individual susceptibility, as well as those affecting biomechanical joint stability [67]. Obesity may also be a risk factor for OA, as it increases stress on weight-bearing joints such as the hips and knees. Additionally, fat cells produce potentially harmful inflammatory proteins that may target the joints. The genetics underlying OA are complex but highly significant and may be related to changes in important molecular pathways. The genes encoding inflammatory and catabolism-related proteins are upregulated during OA, primarily through signal transduction involving nuclear factor-κB (NF-κB), mitogen- activated protein kinase, and other inflammation- and stress-induced pathways [63, 68]. Joint injuries, such as those involving the ligaments and meniscus, can increase the fragility of the joint and increase the risk of osteoarthritis. Even injuries that occurred many years ago can increase the risk of OA. Moreover, joint malformation or cartilage defects can increase the risk of OA [69-71]. The symptoms of OA include pain, transient morning stiffness and a grating sensation when walking. End-stage OA is characterized by instability and physical disability, which have tremendous negative effects on the quality of life [72]. The lack of efficient and curative treatment options for OA and the ever-evolving pathophysiological and risk factors present significant challenges. ...
Incidence of symptomatic hand, hip, and knee osteoarthritis among patients in a health maintenance organization
1
1995
... As noted previously, OA is the most common degenerative joint disorder and can affect nearly any small or large joint in the body [63]. OA is now considered to have complex etiology, and these multiple factors can lead to similar outcomes of joint destruction and clinical features [64]. In a study of OA patients older than 50 years of age in North America and Europe, approximately 60%, 33% and 5% reported involvement of joints of the hand, knee and hip, respectively [65]. An age older than 40 years is associated with a greater likelihood of developing OA. Moreover, OA is more frequent in women than in men at any age older than 50 years (i.e., postmenopausal women), and this sex difference mainly involves the hand (e.g., distal interphalangeal) and knee joints [66]. The risk factors for OA include factors related to individual susceptibility, as well as those affecting biomechanical joint stability [67]. Obesity may also be a risk factor for OA, as it increases stress on weight-bearing joints such as the hips and knees. Additionally, fat cells produce potentially harmful inflammatory proteins that may target the joints. The genetics underlying OA are complex but highly significant and may be related to changes in important molecular pathways. The genes encoding inflammatory and catabolism-related proteins are upregulated during OA, primarily through signal transduction involving nuclear factor-κB (NF-κB), mitogen- activated protein kinase, and other inflammation- and stress-induced pathways [63, 68]. Joint injuries, such as those involving the ligaments and meniscus, can increase the fragility of the joint and increase the risk of osteoarthritis. Even injuries that occurred many years ago can increase the risk of OA. Moreover, joint malformation or cartilage defects can increase the risk of OA [69-71]. The symptoms of OA include pain, transient morning stiffness and a grating sensation when walking. End-stage OA is characterized by instability and physical disability, which have tremendous negative effects on the quality of life [72]. The lack of efficient and curative treatment options for OA and the ever-evolving pathophysiological and risk factors present significant challenges. ...
Risk factors and burden of osteoarthritis
1
2016
... As noted previously, OA is the most common degenerative joint disorder and can affect nearly any small or large joint in the body [63]. OA is now considered to have complex etiology, and these multiple factors can lead to similar outcomes of joint destruction and clinical features [64]. In a study of OA patients older than 50 years of age in North America and Europe, approximately 60%, 33% and 5% reported involvement of joints of the hand, knee and hip, respectively [65]. An age older than 40 years is associated with a greater likelihood of developing OA. Moreover, OA is more frequent in women than in men at any age older than 50 years (i.e., postmenopausal women), and this sex difference mainly involves the hand (e.g., distal interphalangeal) and knee joints [66]. The risk factors for OA include factors related to individual susceptibility, as well as those affecting biomechanical joint stability [67]. Obesity may also be a risk factor for OA, as it increases stress on weight-bearing joints such as the hips and knees. Additionally, fat cells produce potentially harmful inflammatory proteins that may target the joints. The genetics underlying OA are complex but highly significant and may be related to changes in important molecular pathways. The genes encoding inflammatory and catabolism-related proteins are upregulated during OA, primarily through signal transduction involving nuclear factor-κB (NF-κB), mitogen- activated protein kinase, and other inflammation- and stress-induced pathways [63, 68]. Joint injuries, such as those involving the ligaments and meniscus, can increase the fragility of the joint and increase the risk of osteoarthritis. Even injuries that occurred many years ago can increase the risk of OA. Moreover, joint malformation or cartilage defects can increase the risk of OA [69-71]. The symptoms of OA include pain, transient morning stiffness and a grating sensation when walking. End-stage OA is characterized by instability and physical disability, which have tremendous negative effects on the quality of life [72]. The lack of efficient and curative treatment options for OA and the ever-evolving pathophysiological and risk factors present significant challenges. ...
Chaperone-mediated autophagy: roles in disease and aging
1
2014
... As noted previously, OA is the most common degenerative joint disorder and can affect nearly any small or large joint in the body [63]. OA is now considered to have complex etiology, and these multiple factors can lead to similar outcomes of joint destruction and clinical features [64]. In a study of OA patients older than 50 years of age in North America and Europe, approximately 60%, 33% and 5% reported involvement of joints of the hand, knee and hip, respectively [65]. An age older than 40 years is associated with a greater likelihood of developing OA. Moreover, OA is more frequent in women than in men at any age older than 50 years (i.e., postmenopausal women), and this sex difference mainly involves the hand (e.g., distal interphalangeal) and knee joints [66]. The risk factors for OA include factors related to individual susceptibility, as well as those affecting biomechanical joint stability [67]. Obesity may also be a risk factor for OA, as it increases stress on weight-bearing joints such as the hips and knees. Additionally, fat cells produce potentially harmful inflammatory proteins that may target the joints. The genetics underlying OA are complex but highly significant and may be related to changes in important molecular pathways. The genes encoding inflammatory and catabolism-related proteins are upregulated during OA, primarily through signal transduction involving nuclear factor-κB (NF-κB), mitogen- activated protein kinase, and other inflammation- and stress-induced pathways [63, 68]. Joint injuries, such as those involving the ligaments and meniscus, can increase the fragility of the joint and increase the risk of osteoarthritis. Even injuries that occurred many years ago can increase the risk of OA. Moreover, joint malformation or cartilage defects can increase the risk of OA [69-71]. The symptoms of OA include pain, transient morning stiffness and a grating sensation when walking. End-stage OA is characterized by instability and physical disability, which have tremendous negative effects on the quality of life [72]. The lack of efficient and curative treatment options for OA and the ever-evolving pathophysiological and risk factors present significant challenges. ...
Methodologic challenges in studying risk factors for progression of knee osteoarthritis
1
2010
... As noted previously, OA is the most common degenerative joint disorder and can affect nearly any small or large joint in the body [63]. OA is now considered to have complex etiology, and these multiple factors can lead to similar outcomes of joint destruction and clinical features [64]. In a study of OA patients older than 50 years of age in North America and Europe, approximately 60%, 33% and 5% reported involvement of joints of the hand, knee and hip, respectively [65]. An age older than 40 years is associated with a greater likelihood of developing OA. Moreover, OA is more frequent in women than in men at any age older than 50 years (i.e., postmenopausal women), and this sex difference mainly involves the hand (e.g., distal interphalangeal) and knee joints [66]. The risk factors for OA include factors related to individual susceptibility, as well as those affecting biomechanical joint stability [67]. Obesity may also be a risk factor for OA, as it increases stress on weight-bearing joints such as the hips and knees. Additionally, fat cells produce potentially harmful inflammatory proteins that may target the joints. The genetics underlying OA are complex but highly significant and may be related to changes in important molecular pathways. The genes encoding inflammatory and catabolism-related proteins are upregulated during OA, primarily through signal transduction involving nuclear factor-κB (NF-κB), mitogen- activated protein kinase, and other inflammation- and stress-induced pathways [63, 68]. Joint injuries, such as those involving the ligaments and meniscus, can increase the fragility of the joint and increase the risk of osteoarthritis. Even injuries that occurred many years ago can increase the risk of OA. Moreover, joint malformation or cartilage defects can increase the risk of OA [69-71]. The symptoms of OA include pain, transient morning stiffness and a grating sensation when walking. End-stage OA is characterized by instability and physical disability, which have tremendous negative effects on the quality of life [72]. The lack of efficient and curative treatment options for OA and the ever-evolving pathophysiological and risk factors present significant challenges. ...
Osteoarthritis and mortality: A prospective cohort study and systematic review with meta-analysis
0
2016
Risk factors for the incidence and progression of radiographic osteoarthritis of the knee among Japanese
1
2011
... As noted previously, OA is the most common degenerative joint disorder and can affect nearly any small or large joint in the body [63]. OA is now considered to have complex etiology, and these multiple factors can lead to similar outcomes of joint destruction and clinical features [64]. In a study of OA patients older than 50 years of age in North America and Europe, approximately 60%, 33% and 5% reported involvement of joints of the hand, knee and hip, respectively [65]. An age older than 40 years is associated with a greater likelihood of developing OA. Moreover, OA is more frequent in women than in men at any age older than 50 years (i.e., postmenopausal women), and this sex difference mainly involves the hand (e.g., distal interphalangeal) and knee joints [66]. The risk factors for OA include factors related to individual susceptibility, as well as those affecting biomechanical joint stability [67]. Obesity may also be a risk factor for OA, as it increases stress on weight-bearing joints such as the hips and knees. Additionally, fat cells produce potentially harmful inflammatory proteins that may target the joints. The genetics underlying OA are complex but highly significant and may be related to changes in important molecular pathways. The genes encoding inflammatory and catabolism-related proteins are upregulated during OA, primarily through signal transduction involving nuclear factor-κB (NF-κB), mitogen- activated protein kinase, and other inflammation- and stress-induced pathways [63, 68]. Joint injuries, such as those involving the ligaments and meniscus, can increase the fragility of the joint and increase the risk of osteoarthritis. Even injuries that occurred many years ago can increase the risk of OA. Moreover, joint malformation or cartilage defects can increase the risk of OA [69-71]. The symptoms of OA include pain, transient morning stiffness and a grating sensation when walking. End-stage OA is characterized by instability and physical disability, which have tremendous negative effects on the quality of life [72]. The lack of efficient and curative treatment options for OA and the ever-evolving pathophysiological and risk factors present significant challenges. ...
Epidemiology of osteoarthritis: literature update
1
2018
... As noted previously, OA is the most common degenerative joint disorder and can affect nearly any small or large joint in the body [63]. OA is now considered to have complex etiology, and these multiple factors can lead to similar outcomes of joint destruction and clinical features [64]. In a study of OA patients older than 50 years of age in North America and Europe, approximately 60%, 33% and 5% reported involvement of joints of the hand, knee and hip, respectively [65]. An age older than 40 years is associated with a greater likelihood of developing OA. Moreover, OA is more frequent in women than in men at any age older than 50 years (i.e., postmenopausal women), and this sex difference mainly involves the hand (e.g., distal interphalangeal) and knee joints [66]. The risk factors for OA include factors related to individual susceptibility, as well as those affecting biomechanical joint stability [67]. Obesity may also be a risk factor for OA, as it increases stress on weight-bearing joints such as the hips and knees. Additionally, fat cells produce potentially harmful inflammatory proteins that may target the joints. The genetics underlying OA are complex but highly significant and may be related to changes in important molecular pathways. The genes encoding inflammatory and catabolism-related proteins are upregulated during OA, primarily through signal transduction involving nuclear factor-κB (NF-κB), mitogen- activated protein kinase, and other inflammation- and stress-induced pathways [63, 68]. Joint injuries, such as those involving the ligaments and meniscus, can increase the fragility of the joint and increase the risk of osteoarthritis. Even injuries that occurred many years ago can increase the risk of OA. Moreover, joint malformation or cartilage defects can increase the risk of OA [69-71]. The symptoms of OA include pain, transient morning stiffness and a grating sensation when walking. End-stage OA is characterized by instability and physical disability, which have tremendous negative effects on the quality of life [72]. The lack of efficient and curative treatment options for OA and the ever-evolving pathophysiological and risk factors present significant challenges. ...
The role of the cartilage matrix in osteoarthritis
1
2011
... Because its exact pathogenesis has not been fully revealed, OA is characterized as a whole-joint disease associated with pathological changes in all involved tissues. The pathology of OA includes the progressive loss and destruction of cartilage, sclerosis of the subchondral bone, formation of osteophytes, variable degrees of synovial inflammation, degeneration of the ligaments and meniscus and hypertrophy of the whole joint capsule[73, 74]. Initially, the risk factors that cause joint instability, such as ligament injury and excessive body weight, induce the excessive secretion of transforming growth factor β1 (TGF-β1) in the subchondral bone. An excess of this cytokine leads to the uncoupling of bone formation and resorption, which is accompanied by angiogenesis, sensory innervation [21], bone cavity formation and sclerosis [75]. Articular cartilage degeneration is the primary concern in OA [76]. Normally, in an environment with low oxygen, chondrocytes exhibit a low-grade turnover rate to maintain an anabolic-catabolic balance in the cartilage [77]. In OA, however, the expression of genes that encode proteins associated with inflammatory and catabolic responses is upregulated primarily via signal transduction pathways involving NF-κB, mitogen-activated protein kinase and other factors that are activated by inflammation. This transcriptional activity increases the production of primary inflammatory cytokines, such as IL-1β and tumor necrosis factor α (TNF-α), which play critical roles in OA pathology. IL-1β and TNF-α stimulate chondrocytes to release cartilage-degrading enzymes such as metalloproteinases 1, 3 and 13 (MMP1, MMP3 and MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 [78, 79]. These enzymes digest aggrecans and type II collagen to degrade the cartilage matrix. IL-1β and TNF-α also contribute to inflammatory chondrocyte death [80]. Together, these processes lead to a gradual loss of cartilage and the formation of osteochondral fissures, which allow the formation of new endochondral bone accompanied by angiogenesis and sensory innervation [81]. These processes may be closely associated with OA pain. During the late stage of OA, patients lose joint function and present with an immensely narrowed joint space containing little remaining cartilage, especially in the knee and hip joints. ...
Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis
1
2016
... Because its exact pathogenesis has not been fully revealed, OA is characterized as a whole-joint disease associated with pathological changes in all involved tissues. The pathology of OA includes the progressive loss and destruction of cartilage, sclerosis of the subchondral bone, formation of osteophytes, variable degrees of synovial inflammation, degeneration of the ligaments and meniscus and hypertrophy of the whole joint capsule[73, 74]. Initially, the risk factors that cause joint instability, such as ligament injury and excessive body weight, induce the excessive secretion of transforming growth factor β1 (TGF-β1) in the subchondral bone. An excess of this cytokine leads to the uncoupling of bone formation and resorption, which is accompanied by angiogenesis, sensory innervation [21], bone cavity formation and sclerosis [75]. Articular cartilage degeneration is the primary concern in OA [76]. Normally, in an environment with low oxygen, chondrocytes exhibit a low-grade turnover rate to maintain an anabolic-catabolic balance in the cartilage [77]. In OA, however, the expression of genes that encode proteins associated with inflammatory and catabolic responses is upregulated primarily via signal transduction pathways involving NF-κB, mitogen-activated protein kinase and other factors that are activated by inflammation. This transcriptional activity increases the production of primary inflammatory cytokines, such as IL-1β and tumor necrosis factor α (TNF-α), which play critical roles in OA pathology. IL-1β and TNF-α stimulate chondrocytes to release cartilage-degrading enzymes such as metalloproteinases 1, 3 and 13 (MMP1, MMP3 and MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 [78, 79]. These enzymes digest aggrecans and type II collagen to degrade the cartilage matrix. IL-1β and TNF-α also contribute to inflammatory chondrocyte death [80]. Together, these processes lead to a gradual loss of cartilage and the formation of osteochondral fissures, which allow the formation of new endochondral bone accompanied by angiogenesis and sensory innervation [81]. These processes may be closely associated with OA pain. During the late stage of OA, patients lose joint function and present with an immensely narrowed joint space containing little remaining cartilage, especially in the knee and hip joints. ...
Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis
1
2013
... Because its exact pathogenesis has not been fully revealed, OA is characterized as a whole-joint disease associated with pathological changes in all involved tissues. The pathology of OA includes the progressive loss and destruction of cartilage, sclerosis of the subchondral bone, formation of osteophytes, variable degrees of synovial inflammation, degeneration of the ligaments and meniscus and hypertrophy of the whole joint capsule[73, 74]. Initially, the risk factors that cause joint instability, such as ligament injury and excessive body weight, induce the excessive secretion of transforming growth factor β1 (TGF-β1) in the subchondral bone. An excess of this cytokine leads to the uncoupling of bone formation and resorption, which is accompanied by angiogenesis, sensory innervation [21], bone cavity formation and sclerosis [75]. Articular cartilage degeneration is the primary concern in OA [76]. Normally, in an environment with low oxygen, chondrocytes exhibit a low-grade turnover rate to maintain an anabolic-catabolic balance in the cartilage [77]. In OA, however, the expression of genes that encode proteins associated with inflammatory and catabolic responses is upregulated primarily via signal transduction pathways involving NF-κB, mitogen-activated protein kinase and other factors that are activated by inflammation. This transcriptional activity increases the production of primary inflammatory cytokines, such as IL-1β and tumor necrosis factor α (TNF-α), which play critical roles in OA pathology. IL-1β and TNF-α stimulate chondrocytes to release cartilage-degrading enzymes such as metalloproteinases 1, 3 and 13 (MMP1, MMP3 and MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 [78, 79]. These enzymes digest aggrecans and type II collagen to degrade the cartilage matrix. IL-1β and TNF-α also contribute to inflammatory chondrocyte death [80]. Together, these processes lead to a gradual loss of cartilage and the formation of osteochondral fissures, which allow the formation of new endochondral bone accompanied by angiogenesis and sensory innervation [81]. These processes may be closely associated with OA pain. During the late stage of OA, patients lose joint function and present with an immensely narrowed joint space containing little remaining cartilage, especially in the knee and hip joints. ...
Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction
1
2010
... Because its exact pathogenesis has not been fully revealed, OA is characterized as a whole-joint disease associated with pathological changes in all involved tissues. The pathology of OA includes the progressive loss and destruction of cartilage, sclerosis of the subchondral bone, formation of osteophytes, variable degrees of synovial inflammation, degeneration of the ligaments and meniscus and hypertrophy of the whole joint capsule[73, 74]. Initially, the risk factors that cause joint instability, such as ligament injury and excessive body weight, induce the excessive secretion of transforming growth factor β1 (TGF-β1) in the subchondral bone. An excess of this cytokine leads to the uncoupling of bone formation and resorption, which is accompanied by angiogenesis, sensory innervation [21], bone cavity formation and sclerosis [75]. Articular cartilage degeneration is the primary concern in OA [76]. Normally, in an environment with low oxygen, chondrocytes exhibit a low-grade turnover rate to maintain an anabolic-catabolic balance in the cartilage [77]. In OA, however, the expression of genes that encode proteins associated with inflammatory and catabolic responses is upregulated primarily via signal transduction pathways involving NF-κB, mitogen-activated protein kinase and other factors that are activated by inflammation. This transcriptional activity increases the production of primary inflammatory cytokines, such as IL-1β and tumor necrosis factor α (TNF-α), which play critical roles in OA pathology. IL-1β and TNF-α stimulate chondrocytes to release cartilage-degrading enzymes such as metalloproteinases 1, 3 and 13 (MMP1, MMP3 and MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 [78, 79]. These enzymes digest aggrecans and type II collagen to degrade the cartilage matrix. IL-1β and TNF-α also contribute to inflammatory chondrocyte death [80]. Together, these processes lead to a gradual loss of cartilage and the formation of osteochondral fissures, which allow the formation of new endochondral bone accompanied by angiogenesis and sensory innervation [81]. These processes may be closely associated with OA pain. During the late stage of OA, patients lose joint function and present with an immensely narrowed joint space containing little remaining cartilage, especially in the knee and hip joints. ...
Integrins and chondrocyte-matrix interactions in articular cartilage
1
2014
... Because its exact pathogenesis has not been fully revealed, OA is characterized as a whole-joint disease associated with pathological changes in all involved tissues. The pathology of OA includes the progressive loss and destruction of cartilage, sclerosis of the subchondral bone, formation of osteophytes, variable degrees of synovial inflammation, degeneration of the ligaments and meniscus and hypertrophy of the whole joint capsule[73, 74]. Initially, the risk factors that cause joint instability, such as ligament injury and excessive body weight, induce the excessive secretion of transforming growth factor β1 (TGF-β1) in the subchondral bone. An excess of this cytokine leads to the uncoupling of bone formation and resorption, which is accompanied by angiogenesis, sensory innervation [21], bone cavity formation and sclerosis [75]. Articular cartilage degeneration is the primary concern in OA [76]. Normally, in an environment with low oxygen, chondrocytes exhibit a low-grade turnover rate to maintain an anabolic-catabolic balance in the cartilage [77]. In OA, however, the expression of genes that encode proteins associated with inflammatory and catabolic responses is upregulated primarily via signal transduction pathways involving NF-κB, mitogen-activated protein kinase and other factors that are activated by inflammation. This transcriptional activity increases the production of primary inflammatory cytokines, such as IL-1β and tumor necrosis factor α (TNF-α), which play critical roles in OA pathology. IL-1β and TNF-α stimulate chondrocytes to release cartilage-degrading enzymes such as metalloproteinases 1, 3 and 13 (MMP1, MMP3 and MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 [78, 79]. These enzymes digest aggrecans and type II collagen to degrade the cartilage matrix. IL-1β and TNF-α also contribute to inflammatory chondrocyte death [80]. Together, these processes lead to a gradual loss of cartilage and the formation of osteochondral fissures, which allow the formation of new endochondral bone accompanied by angiogenesis and sensory innervation [81]. These processes may be closely associated with OA pain. During the late stage of OA, patients lose joint function and present with an immensely narrowed joint space containing little remaining cartilage, especially in the knee and hip joints. ...
MMP13 is a critical target gene during the progression of osteoarthritis
1
2013
... Because its exact pathogenesis has not been fully revealed, OA is characterized as a whole-joint disease associated with pathological changes in all involved tissues. The pathology of OA includes the progressive loss and destruction of cartilage, sclerosis of the subchondral bone, formation of osteophytes, variable degrees of synovial inflammation, degeneration of the ligaments and meniscus and hypertrophy of the whole joint capsule[73, 74]. Initially, the risk factors that cause joint instability, such as ligament injury and excessive body weight, induce the excessive secretion of transforming growth factor β1 (TGF-β1) in the subchondral bone. An excess of this cytokine leads to the uncoupling of bone formation and resorption, which is accompanied by angiogenesis, sensory innervation [21], bone cavity formation and sclerosis [75]. Articular cartilage degeneration is the primary concern in OA [76]. Normally, in an environment with low oxygen, chondrocytes exhibit a low-grade turnover rate to maintain an anabolic-catabolic balance in the cartilage [77]. In OA, however, the expression of genes that encode proteins associated with inflammatory and catabolic responses is upregulated primarily via signal transduction pathways involving NF-κB, mitogen-activated protein kinase and other factors that are activated by inflammation. This transcriptional activity increases the production of primary inflammatory cytokines, such as IL-1β and tumor necrosis factor α (TNF-α), which play critical roles in OA pathology. IL-1β and TNF-α stimulate chondrocytes to release cartilage-degrading enzymes such as metalloproteinases 1, 3 and 13 (MMP1, MMP3 and MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 [78, 79]. These enzymes digest aggrecans and type II collagen to degrade the cartilage matrix. IL-1β and TNF-α also contribute to inflammatory chondrocyte death [80]. Together, these processes lead to a gradual loss of cartilage and the formation of osteochondral fissures, which allow the formation of new endochondral bone accompanied by angiogenesis and sensory innervation [81]. These processes may be closely associated with OA pain. During the late stage of OA, patients lose joint function and present with an immensely narrowed joint space containing little remaining cartilage, especially in the knee and hip joints. ...
Emerging Frontiers in cartilage and chondrocyte biology
1
2011
... Because its exact pathogenesis has not been fully revealed, OA is characterized as a whole-joint disease associated with pathological changes in all involved tissues. The pathology of OA includes the progressive loss and destruction of cartilage, sclerosis of the subchondral bone, formation of osteophytes, variable degrees of synovial inflammation, degeneration of the ligaments and meniscus and hypertrophy of the whole joint capsule[73, 74]. Initially, the risk factors that cause joint instability, such as ligament injury and excessive body weight, induce the excessive secretion of transforming growth factor β1 (TGF-β1) in the subchondral bone. An excess of this cytokine leads to the uncoupling of bone formation and resorption, which is accompanied by angiogenesis, sensory innervation [21], bone cavity formation and sclerosis [75]. Articular cartilage degeneration is the primary concern in OA [76]. Normally, in an environment with low oxygen, chondrocytes exhibit a low-grade turnover rate to maintain an anabolic-catabolic balance in the cartilage [77]. In OA, however, the expression of genes that encode proteins associated with inflammatory and catabolic responses is upregulated primarily via signal transduction pathways involving NF-κB, mitogen-activated protein kinase and other factors that are activated by inflammation. This transcriptional activity increases the production of primary inflammatory cytokines, such as IL-1β and tumor necrosis factor α (TNF-α), which play critical roles in OA pathology. IL-1β and TNF-α stimulate chondrocytes to release cartilage-degrading enzymes such as metalloproteinases 1, 3 and 13 (MMP1, MMP3 and MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 [78, 79]. These enzymes digest aggrecans and type II collagen to degrade the cartilage matrix. IL-1β and TNF-α also contribute to inflammatory chondrocyte death [80]. Together, these processes lead to a gradual loss of cartilage and the formation of osteochondral fissures, which allow the formation of new endochondral bone accompanied by angiogenesis and sensory innervation [81]. These processes may be closely associated with OA pain. During the late stage of OA, patients lose joint function and present with an immensely narrowed joint space containing little remaining cartilage, especially in the knee and hip joints. ...
Immunopathogenesis of osteoarthritis
1
2013
... Because its exact pathogenesis has not been fully revealed, OA is characterized as a whole-joint disease associated with pathological changes in all involved tissues. The pathology of OA includes the progressive loss and destruction of cartilage, sclerosis of the subchondral bone, formation of osteophytes, variable degrees of synovial inflammation, degeneration of the ligaments and meniscus and hypertrophy of the whole joint capsule[73, 74]. Initially, the risk factors that cause joint instability, such as ligament injury and excessive body weight, induce the excessive secretion of transforming growth factor β1 (TGF-β1) in the subchondral bone. An excess of this cytokine leads to the uncoupling of bone formation and resorption, which is accompanied by angiogenesis, sensory innervation [21], bone cavity formation and sclerosis [75]. Articular cartilage degeneration is the primary concern in OA [76]. Normally, in an environment with low oxygen, chondrocytes exhibit a low-grade turnover rate to maintain an anabolic-catabolic balance in the cartilage [77]. In OA, however, the expression of genes that encode proteins associated with inflammatory and catabolic responses is upregulated primarily via signal transduction pathways involving NF-κB, mitogen-activated protein kinase and other factors that are activated by inflammation. This transcriptional activity increases the production of primary inflammatory cytokines, such as IL-1β and tumor necrosis factor α (TNF-α), which play critical roles in OA pathology. IL-1β and TNF-α stimulate chondrocytes to release cartilage-degrading enzymes such as metalloproteinases 1, 3 and 13 (MMP1, MMP3 and MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 [78, 79]. These enzymes digest aggrecans and type II collagen to degrade the cartilage matrix. IL-1β and TNF-α also contribute to inflammatory chondrocyte death [80]. Together, these processes lead to a gradual loss of cartilage and the formation of osteochondral fissures, which allow the formation of new endochondral bone accompanied by angiogenesis and sensory innervation [81]. These processes may be closely associated with OA pain. During the late stage of OA, patients lose joint function and present with an immensely narrowed joint space containing little remaining cartilage, especially in the knee and hip joints. ...
Anabolic/Catabolic balance in pathogenesis of osteoarthritis: identifying molecular targets
1
2011
... Because its exact pathogenesis has not been fully revealed, OA is characterized as a whole-joint disease associated with pathological changes in all involved tissues. The pathology of OA includes the progressive loss and destruction of cartilage, sclerosis of the subchondral bone, formation of osteophytes, variable degrees of synovial inflammation, degeneration of the ligaments and meniscus and hypertrophy of the whole joint capsule[73, 74]. Initially, the risk factors that cause joint instability, such as ligament injury and excessive body weight, induce the excessive secretion of transforming growth factor β1 (TGF-β1) in the subchondral bone. An excess of this cytokine leads to the uncoupling of bone formation and resorption, which is accompanied by angiogenesis, sensory innervation [21], bone cavity formation and sclerosis [75]. Articular cartilage degeneration is the primary concern in OA [76]. Normally, in an environment with low oxygen, chondrocytes exhibit a low-grade turnover rate to maintain an anabolic-catabolic balance in the cartilage [77]. In OA, however, the expression of genes that encode proteins associated with inflammatory and catabolic responses is upregulated primarily via signal transduction pathways involving NF-κB, mitogen-activated protein kinase and other factors that are activated by inflammation. This transcriptional activity increases the production of primary inflammatory cytokines, such as IL-1β and tumor necrosis factor α (TNF-α), which play critical roles in OA pathology. IL-1β and TNF-α stimulate chondrocytes to release cartilage-degrading enzymes such as metalloproteinases 1, 3 and 13 (MMP1, MMP3 and MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4) and ADAMTS5 [78, 79]. These enzymes digest aggrecans and type II collagen to degrade the cartilage matrix. IL-1β and TNF-α also contribute to inflammatory chondrocyte death [80]. Together, these processes lead to a gradual loss of cartilage and the formation of osteochondral fissures, which allow the formation of new endochondral bone accompanied by angiogenesis and sensory innervation [81]. These processes may be closely associated with OA pain. During the late stage of OA, patients lose joint function and present with an immensely narrowed joint space containing little remaining cartilage, especially in the knee and hip joints. ...
The role of synovitis in osteoarthritis pathogenesis
1
2012
... The synovium is an important source of nutrients for cartilaginous tissue, which is avascular. Therefore, synovitis, or inflammation of the synovium, plays a key role in OA pathogenesis [82]. The synovium includes the synovial membrane and fluid [83]. Damaged chondrocytes and cartilage release proteinases that increase the concentrations of proinflammatory cartilage degradation products, such as DAMPs, in the synovial fluid. These products induce inflammation in the adjacent synovium [84, 85] and present feedback to the chondrocytes for further degradation. Consequently, increased amounts of matrix-degrading enzymes are secreted to deteriorate the cartilage, and reduced amounts of lubricin and hyaluronic acid (HA) are secreted to protect the joints [86]. ...
Synovitis and the risk of knee osteoarthritis: the MOST Study
1
2016
... The synovium is an important source of nutrients for cartilaginous tissue, which is avascular. Therefore, synovitis, or inflammation of the synovium, plays a key role in OA pathogenesis [82]. The synovium includes the synovial membrane and fluid [83]. Damaged chondrocytes and cartilage release proteinases that increase the concentrations of proinflammatory cartilage degradation products, such as DAMPs, in the synovial fluid. These products induce inflammation in the adjacent synovium [84, 85] and present feedback to the chondrocytes for further degradation. Consequently, increased amounts of matrix-degrading enzymes are secreted to deteriorate the cartilage, and reduced amounts of lubricin and hyaluronic acid (HA) are secreted to protect the joints [86]. ...
Osteoarthritis: a disease of the joint as an organ
1
2012
... The synovium is an important source of nutrients for cartilaginous tissue, which is avascular. Therefore, synovitis, or inflammation of the synovium, plays a key role in OA pathogenesis [82]. The synovium includes the synovial membrane and fluid [83]. Damaged chondrocytes and cartilage release proteinases that increase the concentrations of proinflammatory cartilage degradation products, such as DAMPs, in the synovial fluid. These products induce inflammation in the adjacent synovium [84, 85] and present feedback to the chondrocytes for further degradation. Consequently, increased amounts of matrix-degrading enzymes are secreted to deteriorate the cartilage, and reduced amounts of lubricin and hyaluronic acid (HA) are secreted to protect the joints [86]. ...
Emerging regulators of the inflammatory process in osteoarthritis
1
2015
... The synovium is an important source of nutrients for cartilaginous tissue, which is avascular. Therefore, synovitis, or inflammation of the synovium, plays a key role in OA pathogenesis [82]. The synovium includes the synovial membrane and fluid [83]. Damaged chondrocytes and cartilage release proteinases that increase the concentrations of proinflammatory cartilage degradation products, such as DAMPs, in the synovial fluid. These products induce inflammation in the adjacent synovium [84, 85] and present feedback to the chondrocytes for further degradation. Consequently, increased amounts of matrix-degrading enzymes are secreted to deteriorate the cartilage, and reduced amounts of lubricin and hyaluronic acid (HA) are secreted to protect the joints [86]. ...
Synovitis in osteoarthritis: current understanding with therapeutic implications
1
2017
... The synovium is an important source of nutrients for cartilaginous tissue, which is avascular. Therefore, synovitis, or inflammation of the synovium, plays a key role in OA pathogenesis [82]. The synovium includes the synovial membrane and fluid [83]. Damaged chondrocytes and cartilage release proteinases that increase the concentrations of proinflammatory cartilage degradation products, such as DAMPs, in the synovial fluid. These products induce inflammation in the adjacent synovium [84, 85] and present feedback to the chondrocytes for further degradation. Consequently, increased amounts of matrix-degrading enzymes are secreted to deteriorate the cartilage, and reduced amounts of lubricin and hyaluronic acid (HA) are secreted to protect the joints [86]. ...
Prediction of long-term mortality in patients with rheumatoid arthritis according to simple questionnaire and joint count measures
2
1994
... To date, a limited amount of literature has described the role of the NLRP3 inflammasome in OA pathogenesis. Since strong evidence shows that NLRP3 is a potential marker in other diseases such as RA [87], atherosclerosis [88], gout [89] and colorectal cancer [90], we detailed the evidence that indicates the association of NLRP3 with OA in the following sections. ...
... Chronic synovial inflammation is a known cause of cartilage degradation [87, 107]. In OA, IL-1β, which is involved in cartilage degradation, may be produced by synovial tissue rather than by chondrocytes [108]. Furthermore, the protein level of NLRP3 is more than five-fold higher in the synovial membranes of OA patients than in subjects with normal joints [109]. Genetic studies have proven a close association of inflammasomes with RA, which is a primary autoimmune disease of the synovium. These studies revealed the significantly upregulated expression of genes encoding NLRP3 inflammasome components, including NLRP3, ASC and caspase-1, in patients with RA [62, 110]. ...
Pharmacological Effects and Mechanisms of Chinese Medicines Modulating NLRP3 Inflammasomes in Ischemic Cardio/Cerebral Vascular Disease
1
2018
... To date, a limited amount of literature has described the role of the NLRP3 inflammasome in OA pathogenesis. Since strong evidence shows that NLRP3 is a potential marker in other diseases such as RA [87], atherosclerosis [88], gout [89] and colorectal cancer [90], we detailed the evidence that indicates the association of NLRP3 with OA in the following sections. ...
Novel insights into the role of inflammasomes in autoimmune and metabolic rheumatic diseases
1
2018
... To date, a limited amount of literature has described the role of the NLRP3 inflammasome in OA pathogenesis. Since strong evidence shows that NLRP3 is a potential marker in other diseases such as RA [87], atherosclerosis [88], gout [89] and colorectal cancer [90], we detailed the evidence that indicates the association of NLRP3 with OA in the following sections. ...
NLRP3 Inflammasome: A Possible Link Between Obesity-Associated Low-Grade Chronic Inflammation and Colorectal Cancer Development
1
2018
... To date, a limited amount of literature has described the role of the NLRP3 inflammasome in OA pathogenesis. Since strong evidence shows that NLRP3 is a potential marker in other diseases such as RA [87], atherosclerosis [88], gout [89] and colorectal cancer [90], we detailed the evidence that indicates the association of NLRP3 with OA in the following sections. ...
Risk factors for onset of osteoarthritis of the knee in older adults: a systematic review and meta-analysis
1
2010
... Metabolic disorders contribute to OA pathogenesis. Obesity (body mass index > 30 kg/m2) is a significant risk factor for the onset and progression of OA [91]. Waist circumference, which is an indicator of central adiposity, is associated with increased knee cartilage defects [92], reduced knee cartilage mass and increased bone marrow lesions [93]. Adipokines, one of the obesity-related metabolic factors, have been shown to induce cartilage degradation [64]. Leptin, an adipokine, has a detrimental effect on chondrocyte proliferation by increasing IL-1β and MMPs [94]. Additionally, individuals with obesity have low circulating 25-hydroxy-vitamin D (25-(OH)D) [95], which is positively associated with the development and worsening of knee OA, including cartilage loss and increased OA pain [96]. ...
The longitudinal relationship between body composition and patella cartilage in healthy adults
1
2008
... Metabolic disorders contribute to OA pathogenesis. Obesity (body mass index > 30 kg/m2) is a significant risk factor for the onset and progression of OA [91]. Waist circumference, which is an indicator of central adiposity, is associated with increased knee cartilage defects [92], reduced knee cartilage mass and increased bone marrow lesions [93]. Adipokines, one of the obesity-related metabolic factors, have been shown to induce cartilage degradation [64]. Leptin, an adipokine, has a detrimental effect on chondrocyte proliferation by increasing IL-1β and MMPs [94]. Additionally, individuals with obesity have low circulating 25-hydroxy-vitamin D (25-(OH)D) [95], which is positively associated with the development and worsening of knee OA, including cartilage loss and increased OA pain [96]. ...
The relationship between body composition and structural changes at the knee
1
2010
... Metabolic disorders contribute to OA pathogenesis. Obesity (body mass index > 30 kg/m2) is a significant risk factor for the onset and progression of OA [91]. Waist circumference, which is an indicator of central adiposity, is associated with increased knee cartilage defects [92], reduced knee cartilage mass and increased bone marrow lesions [93]. Adipokines, one of the obesity-related metabolic factors, have been shown to induce cartilage degradation [64]. Leptin, an adipokine, has a detrimental effect on chondrocyte proliferation by increasing IL-1β and MMPs [94]. Additionally, individuals with obesity have low circulating 25-hydroxy-vitamin D (25-(OH)D) [95], which is positively associated with the development and worsening of knee OA, including cartilage loss and increased OA pain [96]. ...
Leptin plays a catabolic role on articular cartilage
1
2010
... Metabolic disorders contribute to OA pathogenesis. Obesity (body mass index > 30 kg/m2) is a significant risk factor for the onset and progression of OA [91]. Waist circumference, which is an indicator of central adiposity, is associated with increased knee cartilage defects [92], reduced knee cartilage mass and increased bone marrow lesions [93]. Adipokines, one of the obesity-related metabolic factors, have been shown to induce cartilage degradation [64]. Leptin, an adipokine, has a detrimental effect on chondrocyte proliferation by increasing IL-1β and MMPs [94]. Additionally, individuals with obesity have low circulating 25-hydroxy-vitamin D (25-(OH)D) [95], which is positively associated with the development and worsening of knee OA, including cartilage loss and increased OA pain [96]. ...
Relation of body fat indexes to vitamin D status and deficiency among obese adolescents
2
2009
... Metabolic disorders contribute to OA pathogenesis. Obesity (body mass index > 30 kg/m2) is a significant risk factor for the onset and progression of OA [91]. Waist circumference, which is an indicator of central adiposity, is associated with increased knee cartilage defects [92], reduced knee cartilage mass and increased bone marrow lesions [93]. Adipokines, one of the obesity-related metabolic factors, have been shown to induce cartilage degradation [64]. Leptin, an adipokine, has a detrimental effect on chondrocyte proliferation by increasing IL-1β and MMPs [94]. Additionally, individuals with obesity have low circulating 25-hydroxy-vitamin D (25-(OH)D) [95], which is positively associated with the development and worsening of knee OA, including cartilage loss and increased OA pain [96]. ...
... Adipose tissue macrophages (ATMs) exhibit a proinflammatory M1 phenotype under obese conditions but an M2-dominant phenotype under normal conditions. Proinflammatory ATMs produce tumor-promoting cytokines, including TNF-α, IL-1β, IL-6, IL-8, IL-18 and IL-32 [95], and therefore serve as a source of both local and systemic proinflammatory mediators [96]. Factors such as oxidized low-density lipoprotein (LDL) and cholesterol can activate the NLRP3 inflammasome. Vandanmagsar et al [97] found that ablation of NLRP3 in mice prevented obesity-induced inflammasome activation in fat deposits. In other words, obesity promotes pyroptosis by enabling the release of the NLRP3 inflammasome, which may be related to OA progression. Furthermore, humans with obesity have elevated blood LPS levels. LPS activates caspase-4/5/11, which initiates pyroptosis via the noncanonical inflammasome pathway [99] at both the systemic and tissue levels, leading to increased inflammatory chondrocyte death and loss of cartilage. ...
Moderate vitamin D deficiency is associated with changes in knee and hip pain in older adults: a 5-year longitudinal study
2
2014
... Metabolic disorders contribute to OA pathogenesis. Obesity (body mass index > 30 kg/m2) is a significant risk factor for the onset and progression of OA [91]. Waist circumference, which is an indicator of central adiposity, is associated with increased knee cartilage defects [92], reduced knee cartilage mass and increased bone marrow lesions [93]. Adipokines, one of the obesity-related metabolic factors, have been shown to induce cartilage degradation [64]. Leptin, an adipokine, has a detrimental effect on chondrocyte proliferation by increasing IL-1β and MMPs [94]. Additionally, individuals with obesity have low circulating 25-hydroxy-vitamin D (25-(OH)D) [95], which is positively associated with the development and worsening of knee OA, including cartilage loss and increased OA pain [96]. ...
... Adipose tissue macrophages (ATMs) exhibit a proinflammatory M1 phenotype under obese conditions but an M2-dominant phenotype under normal conditions. Proinflammatory ATMs produce tumor-promoting cytokines, including TNF-α, IL-1β, IL-6, IL-8, IL-18 and IL-32 [95], and therefore serve as a source of both local and systemic proinflammatory mediators [96]. Factors such as oxidized low-density lipoprotein (LDL) and cholesterol can activate the NLRP3 inflammasome. Vandanmagsar et al [97] found that ablation of NLRP3 in mice prevented obesity-induced inflammasome activation in fat deposits. In other words, obesity promotes pyroptosis by enabling the release of the NLRP3 inflammasome, which may be related to OA progression. Furthermore, humans with obesity have elevated blood LPS levels. LPS activates caspase-4/5/11, which initiates pyroptosis via the noncanonical inflammasome pathway [99] at both the systemic and tissue levels, leading to increased inflammatory chondrocyte death and loss of cartilage. ...
The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance
1
2011
... Adipose tissue macrophages (ATMs) exhibit a proinflammatory M1 phenotype under obese conditions but an M2-dominant phenotype under normal conditions. Proinflammatory ATMs produce tumor-promoting cytokines, including TNF-α, IL-1β, IL-6, IL-8, IL-18 and IL-32 [95], and therefore serve as a source of both local and systemic proinflammatory mediators [96]. Factors such as oxidized low-density lipoprotein (LDL) and cholesterol can activate the NLRP3 inflammasome. Vandanmagsar et al [97] found that ablation of NLRP3 in mice prevented obesity-induced inflammasome activation in fat deposits. In other words, obesity promotes pyroptosis by enabling the release of the NLRP3 inflammasome, which may be related to OA progression. Furthermore, humans with obesity have elevated blood LPS levels. LPS activates caspase-4/5/11, which initiates pyroptosis via the noncanonical inflammasome pathway [99] at both the systemic and tissue levels, leading to increased inflammatory chondrocyte death and loss of cartilage. ...
Microcrystals as DAMPs and their role in joint inflammation
1
2012
... Microcrystals, including basic calcium phosphate (BCP), calcium pyrophosphate and uric acid [98], are considered “danger signals” and are thought to be the main DAMPs that activate inflammasomes. BCP crystals have been found in the knee and hip joints of OA patients [99], and these crystals elicit the release of IL-1β from macrophages by activating NLRP3 in a process that is dependent on K+ efflux, reactive oxygen species (ROS) and phagocytosis [100, 101]. This pyroptosis-related process drives the pathology of OA and induces cartilage degeneration. Previous reports[102] suggested that in synovial fibroblasts and articular chondrocytes, IL-36, an IL-1 cytokine subfamily member, stimulates the expression of inflammatory mediators and thus plays a critical role in inflammatory diseases [59]. ...
Osteoarthritis-associated basic calcium phosphate crystals activate membrane proximal kinases in human innate immune cells
2
2017
... Adipose tissue macrophages (ATMs) exhibit a proinflammatory M1 phenotype under obese conditions but an M2-dominant phenotype under normal conditions. Proinflammatory ATMs produce tumor-promoting cytokines, including TNF-α, IL-1β, IL-6, IL-8, IL-18 and IL-32 [95], and therefore serve as a source of both local and systemic proinflammatory mediators [96]. Factors such as oxidized low-density lipoprotein (LDL) and cholesterol can activate the NLRP3 inflammasome. Vandanmagsar et al [97] found that ablation of NLRP3 in mice prevented obesity-induced inflammasome activation in fat deposits. In other words, obesity promotes pyroptosis by enabling the release of the NLRP3 inflammasome, which may be related to OA progression. Furthermore, humans with obesity have elevated blood LPS levels. LPS activates caspase-4/5/11, which initiates pyroptosis via the noncanonical inflammasome pathway [99] at both the systemic and tissue levels, leading to increased inflammatory chondrocyte death and loss of cartilage. ...
... Microcrystals, including basic calcium phosphate (BCP), calcium pyrophosphate and uric acid [98], are considered “danger signals” and are thought to be the main DAMPs that activate inflammasomes. BCP crystals have been found in the knee and hip joints of OA patients [99], and these crystals elicit the release of IL-1β from macrophages by activating NLRP3 in a process that is dependent on K+ efflux, reactive oxygen species (ROS) and phagocytosis [100, 101]. This pyroptosis-related process drives the pathology of OA and induces cartilage degeneration. Previous reports[102] suggested that in synovial fibroblasts and articular chondrocytes, IL-36, an IL-1 cytokine subfamily member, stimulates the expression of inflammatory mediators and thus plays a critical role in inflammatory diseases [59]. ...
NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy
1
2011
... Microcrystals, including basic calcium phosphate (BCP), calcium pyrophosphate and uric acid [98], are considered “danger signals” and are thought to be the main DAMPs that activate inflammasomes. BCP crystals have been found in the knee and hip joints of OA patients [99], and these crystals elicit the release of IL-1β from macrophages by activating NLRP3 in a process that is dependent on K+ efflux, reactive oxygen species (ROS) and phagocytosis [100, 101]. This pyroptosis-related process drives the pathology of OA and induces cartilage degeneration. Previous reports[102] suggested that in synovial fibroblasts and articular chondrocytes, IL-36, an IL-1 cytokine subfamily member, stimulates the expression of inflammatory mediators and thus plays a critical role in inflammatory diseases [59]. ...
Basic calcium phosphate crystals induce monocyte/macrophage IL-1beta secretion through the NLRP3 inflammasome in vitro
1
2011
... Microcrystals, including basic calcium phosphate (BCP), calcium pyrophosphate and uric acid [98], are considered “danger signals” and are thought to be the main DAMPs that activate inflammasomes. BCP crystals have been found in the knee and hip joints of OA patients [99], and these crystals elicit the release of IL-1β from macrophages by activating NLRP3 in a process that is dependent on K+ efflux, reactive oxygen species (ROS) and phagocytosis [100, 101]. This pyroptosis-related process drives the pathology of OA and induces cartilage degeneration. Previous reports[102] suggested that in synovial fibroblasts and articular chondrocytes, IL-36, an IL-1 cytokine subfamily member, stimulates the expression of inflammatory mediators and thus plays a critical role in inflammatory diseases [59]. ...
TGFRII mediated modulation of IL-36 for OA
1
... Microcrystals, including basic calcium phosphate (BCP), calcium pyrophosphate and uric acid [98], are considered “danger signals” and are thought to be the main DAMPs that activate inflammasomes. BCP crystals have been found in the knee and hip joints of OA patients [99], and these crystals elicit the release of IL-1β from macrophages by activating NLRP3 in a process that is dependent on K+ efflux, reactive oxygen species (ROS) and phagocytosis [100, 101]. This pyroptosis-related process drives the pathology of OA and induces cartilage degeneration. Previous reports[102] suggested that in synovial fibroblasts and articular chondrocytes, IL-36, an IL-1 cytokine subfamily member, stimulates the expression of inflammatory mediators and thus plays a critical role in inflammatory diseases [59]. ...
Drivers of age-related inflammation and strategies for healthspan extension
1
2015
... Aging is also a significant risk factor for multiple chronic diseases, as elderly people are more vulnerable due to their decreased immune and metabolic activities. A highly critical event occurs with aging, which starts with DAMP accumulation and activation of the subsequent NLRP3 inflammasome, which is stimulated by endogenous byproducts [103] that are then recognized by PRRs in macrophages to trigger chronic, low-grade inflammation [31]. In turn, chronic inflammation plays an important role in the progression of metabolic disorders, such as obesity, gouty arthritis and atherosclerosis [104]. ...
Growth Hormone Receptor Deficiency Protects against Age-Related NLRP3 Inflammasome Activation and Immune Senescence
1
2016
... Aging is also a significant risk factor for multiple chronic diseases, as elderly people are more vulnerable due to their decreased immune and metabolic activities. A highly critical event occurs with aging, which starts with DAMP accumulation and activation of the subsequent NLRP3 inflammasome, which is stimulated by endogenous byproducts [103] that are then recognized by PRRs in macrophages to trigger chronic, low-grade inflammation [31]. In turn, chronic inflammation plays an important role in the progression of metabolic disorders, such as obesity, gouty arthritis and atherosclerosis [104]. ...
Regulation of inflammasome activation
1
2015
... Upon stimulation by DAMPs, macrophages surrounding the cartilage undergo pyroptosis after caspase-1 activation by DAMPs or PAMPs and the release inflammasomes such as NLRP3. This increases the release of IL-1β and IL-18 on the cartilage surface, leading to increases in the concentrations of proinflammatory cytokines in chondrocytes and further promoting pyroptosis [105]. These cytokines stimulate chondrocytes to secrete catabolic enzymes that cause cartilage degradation, such as MMP13 and ADAMTS5 [106], thus leading to cartilage destruction. ...
Hydroxyurea for children with sickle cell disease: are we starting too late?
1
2011
... Upon stimulation by DAMPs, macrophages surrounding the cartilage undergo pyroptosis after caspase-1 activation by DAMPs or PAMPs and the release inflammasomes such as NLRP3. This increases the release of IL-1β and IL-18 on the cartilage surface, leading to increases in the concentrations of proinflammatory cytokines in chondrocytes and further promoting pyroptosis [105]. These cytokines stimulate chondrocytes to secrete catabolic enzymes that cause cartilage degradation, such as MMP13 and ADAMTS5 [106], thus leading to cartilage destruction. ...
Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I
1
2008
... Chronic synovial inflammation is a known cause of cartilage degradation [87, 107]. In OA, IL-1β, which is involved in cartilage degradation, may be produced by synovial tissue rather than by chondrocytes [108]. Furthermore, the protein level of NLRP3 is more than five-fold higher in the synovial membranes of OA patients than in subjects with normal joints [109]. Genetic studies have proven a close association of inflammasomes with RA, which is a primary autoimmune disease of the synovium. These studies revealed the significantly upregulated expression of genes encoding NLRP3 inflammasome components, including NLRP3, ASC and caspase-1, in patients with RA [62, 110]. ...
Altered expression of circulating microRNA in plasma of patients with primary osteoarthritis and in silico analysis of their pathways
1
2014
... Chronic synovial inflammation is a known cause of cartilage degradation [87, 107]. In OA, IL-1β, which is involved in cartilage degradation, may be produced by synovial tissue rather than by chondrocytes [108]. Furthermore, the protein level of NLRP3 is more than five-fold higher in the synovial membranes of OA patients than in subjects with normal joints [109]. Genetic studies have proven a close association of inflammasomes with RA, which is a primary autoimmune disease of the synovium. These studies revealed the significantly upregulated expression of genes encoding NLRP3 inflammasome components, including NLRP3, ASC and caspase-1, in patients with RA [62, 110]. ...
The Overexpression of NALP3 Inflammasome in Knee Osteoarthritis Is Associated with Synovial Membrane Prolidase and NADPH Oxidase 2
1
2016
... Chronic synovial inflammation is a known cause of cartilage degradation [87, 107]. In OA, IL-1β, which is involved in cartilage degradation, may be produced by synovial tissue rather than by chondrocytes [108]. Furthermore, the protein level of NLRP3 is more than five-fold higher in the synovial membranes of OA patients than in subjects with normal joints [109]. Genetic studies have proven a close association of inflammasomes with RA, which is a primary autoimmune disease of the synovium. These studies revealed the significantly upregulated expression of genes encoding NLRP3 inflammasome components, including NLRP3, ASC and caspase-1, in patients with RA [62, 110]. ...
NLRP3 and CARD8 Polymorphisms Influence Higher Disease Activity in Rheumatoid Arthritis
1
2016
... Chronic synovial inflammation is a known cause of cartilage degradation [87, 107]. In OA, IL-1β, which is involved in cartilage degradation, may be produced by synovial tissue rather than by chondrocytes [108]. Furthermore, the protein level of NLRP3 is more than five-fold higher in the synovial membranes of OA patients than in subjects with normal joints [109]. Genetic studies have proven a close association of inflammasomes with RA, which is a primary autoimmune disease of the synovium. These studies revealed the significantly upregulated expression of genes encoding NLRP3 inflammasome components, including NLRP3, ASC and caspase-1, in patients with RA [62, 110]. ...
Stress-induced cartilage degradation does not depend on the NLRP3 inflammasome in human osteoarthritis and mouse models
2
2012
... In patients with knee OA, the level of uric acid, a danger signal, in the synovial fluid was found to correlate with the synovial levels of IL-1β and IL-18 and the severity of OA [111]. Therefore, uric acid is a driver of OA pathogenesis and may be a marker of OA severity [112]. Chondrocytes produce increased IL-1β and IL-18, while IL-1β also independently induces the expression of IL-18, which enhances the local inflammatory activities independently in the surrounding synovial fluid [113], enhancing local inflammation during the progression of OA. ...
... However, our understanding of the role of pyroptosis in OA remains limited. Moreover, there were also some opposing data that do not support the role of pyroptosis in OA. Busso et al [118] found in a murine model that IL-1α and IL-1β were not key mediators in OA. A lack of IL-1α or NLRP3 could lead to increased cartilage erosion. Bougault et al [111] found that knockout of NLRP3 did not inhibit the expression of MMP3, MMP9 and MMP13 in cultured chondrocytes, and this was no changed by inhibiting caspase-1 or IL-1β. These studies all indicated that inhibiting IL-1β was not a promising therapeutic target, which was consistent with human clinical trials[119]. However, these data do not necessarily disprove the role of pyroptosis in OA. First, the animal and cartilage explant models in these studies may not accurately reflect the aspects of the disease that are mediated by pyroptosis. Second, pyroptosis may be a process that is closely related to cell death, not just inflammatory factors such as IL-1β. ...
Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation
1
2011
... In patients with knee OA, the level of uric acid, a danger signal, in the synovial fluid was found to correlate with the synovial levels of IL-1β and IL-18 and the severity of OA [111]. Therefore, uric acid is a driver of OA pathogenesis and may be a marker of OA severity [112]. Chondrocytes produce increased IL-1β and IL-18, while IL-1β also independently induces the expression of IL-18, which enhances the local inflammatory activities independently in the surrounding synovial fluid [113], enhancing local inflammation during the progression of OA. ...
IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses
1
1999
... In patients with knee OA, the level of uric acid, a danger signal, in the synovial fluid was found to correlate with the synovial levels of IL-1β and IL-18 and the severity of OA [111]. Therefore, uric acid is a driver of OA pathogenesis and may be a marker of OA severity [112]. Chondrocytes produce increased IL-1β and IL-18, while IL-1β also independently induces the expression of IL-18, which enhances the local inflammatory activities independently in the surrounding synovial fluid [113], enhancing local inflammation during the progression of OA. ...
Lipid mediators of sensitivity in sensory neurons
1
2005
... Pyroptosis may also contribute to the pathological mechanism underlying pain, which is one of the most characteristic symptoms of OA. Continuous nociceptive input from the OA joint is thought to sensitize central and peripheral nerves and induce pain [114]. The majority of OA pain is caused by changes in peripheral nerves. Normally, nerve nociceptors are localized within specific tissues and can only detect sensations in a designated area. During OA progression, excess cytokines, chemokines and inflammatory factors, mechanical stimuli and enhanced innervation contribute to the expansion of nociceptive input [115]. In OA pathology, inflammasomes in pyroptotic macrophages upregulate the production of proinflammatory cytokines, such as IL-1β, IL-18 and TNF-α, which can increase nociceptive input in the joint tissue. This process is called “hyperalgesia.” Additionally, in the context of chronic inflammation associated with OA, pyroptosis may contribute to pathological changes in the whole joint, including the loss of cartilage, formation of osteochondral fissures and osteophytes and synovitis [116]. Furthermore, dense networks of perivascular sensory and sympathetic nerves may innervate the channels or extend into the synovium, ligament or meniscus [116, 117]. This abnormal pattern of innervation would surely contribute to OA pain. ...
Cytokine and chemokine regulation of sensory neuron function
1
2009
... Pyroptosis may also contribute to the pathological mechanism underlying pain, which is one of the most characteristic symptoms of OA. Continuous nociceptive input from the OA joint is thought to sensitize central and peripheral nerves and induce pain [114]. The majority of OA pain is caused by changes in peripheral nerves. Normally, nerve nociceptors are localized within specific tissues and can only detect sensations in a designated area. During OA progression, excess cytokines, chemokines and inflammatory factors, mechanical stimuli and enhanced innervation contribute to the expansion of nociceptive input [115]. In OA pathology, inflammasomes in pyroptotic macrophages upregulate the production of proinflammatory cytokines, such as IL-1β, IL-18 and TNF-α, which can increase nociceptive input in the joint tissue. This process is called “hyperalgesia.” Additionally, in the context of chronic inflammation associated with OA, pyroptosis may contribute to pathological changes in the whole joint, including the loss of cartilage, formation of osteochondral fissures and osteophytes and synovitis [116]. Furthermore, dense networks of perivascular sensory and sympathetic nerves may innervate the channels or extend into the synovium, ligament or meniscus [116, 117]. This abnormal pattern of innervation would surely contribute to OA pain. ...
Mechanisms and targets of angiogenesis and nerve growth in osteoarthritis
2
2012
... Pyroptosis may also contribute to the pathological mechanism underlying pain, which is one of the most characteristic symptoms of OA. Continuous nociceptive input from the OA joint is thought to sensitize central and peripheral nerves and induce pain [114]. The majority of OA pain is caused by changes in peripheral nerves. Normally, nerve nociceptors are localized within specific tissues and can only detect sensations in a designated area. During OA progression, excess cytokines, chemokines and inflammatory factors, mechanical stimuli and enhanced innervation contribute to the expansion of nociceptive input [115]. In OA pathology, inflammasomes in pyroptotic macrophages upregulate the production of proinflammatory cytokines, such as IL-1β, IL-18 and TNF-α, which can increase nociceptive input in the joint tissue. This process is called “hyperalgesia.” Additionally, in the context of chronic inflammation associated with OA, pyroptosis may contribute to pathological changes in the whole joint, including the loss of cartilage, formation of osteochondral fissures and osteophytes and synovitis [116]. Furthermore, dense networks of perivascular sensory and sympathetic nerves may innervate the channels or extend into the synovium, ligament or meniscus [116, 117]. This abnormal pattern of innervation would surely contribute to OA pain. ...
... ]. Furthermore, dense networks of perivascular sensory and sympathetic nerves may innervate the channels or extend into the synovium, ligament or meniscus [116, 117]. This abnormal pattern of innervation would surely contribute to OA pain. ...
Anatomy and histology of joint innervation
1
1997
... Pyroptosis may also contribute to the pathological mechanism underlying pain, which is one of the most characteristic symptoms of OA. Continuous nociceptive input from the OA joint is thought to sensitize central and peripheral nerves and induce pain [114]. The majority of OA pain is caused by changes in peripheral nerves. Normally, nerve nociceptors are localized within specific tissues and can only detect sensations in a designated area. During OA progression, excess cytokines, chemokines and inflammatory factors, mechanical stimuli and enhanced innervation contribute to the expansion of nociceptive input [115]. In OA pathology, inflammasomes in pyroptotic macrophages upregulate the production of proinflammatory cytokines, such as IL-1β, IL-18 and TNF-α, which can increase nociceptive input in the joint tissue. This process is called “hyperalgesia.” Additionally, in the context of chronic inflammation associated with OA, pyroptosis may contribute to pathological changes in the whole joint, including the loss of cartilage, formation of osteochondral fissures and osteophytes and synovitis [116]. Furthermore, dense networks of perivascular sensory and sympathetic nerves may innervate the channels or extend into the synovium, ligament or meniscus [116, 117]. This abnormal pattern of innervation would surely contribute to OA pain. ...
Revisiting the Role of Interleukin-1 Pathway in Osteoarthritis: Interleukin-1alpha and -1beta, and NLRP3 Inflammasome Are Not Involved in the Pathological Features of the Murine Menisectomy Model of Osteoarthritis
1
2017
... However, our understanding of the role of pyroptosis in OA remains limited. Moreover, there were also some opposing data that do not support the role of pyroptosis in OA. Busso et al [118] found in a murine model that IL-1α and IL-1β were not key mediators in OA. A lack of IL-1α or NLRP3 could lead to increased cartilage erosion. Bougault et al [111] found that knockout of NLRP3 did not inhibit the expression of MMP3, MMP9 and MMP13 in cultured chondrocytes, and this was no changed by inhibiting caspase-1 or IL-1β. These studies all indicated that inhibiting IL-1β was not a promising therapeutic target, which was consistent with human clinical trials[119]. However, these data do not necessarily disprove the role of pyroptosis in OA. First, the animal and cartilage explant models in these studies may not accurately reflect the aspects of the disease that are mediated by pyroptosis. Second, pyroptosis may be a process that is closely related to cell death, not just inflammatory factors such as IL-1β. ...
IL-1 in osteoarthritis: time for a critical review of the literature
1
2019
... However, our understanding of the role of pyroptosis in OA remains limited. Moreover, there were also some opposing data that do not support the role of pyroptosis in OA. Busso et al [118] found in a murine model that IL-1α and IL-1β were not key mediators in OA. A lack of IL-1α or NLRP3 could lead to increased cartilage erosion. Bougault et al [111] found that knockout of NLRP3 did not inhibit the expression of MMP3, MMP9 and MMP13 in cultured chondrocytes, and this was no changed by inhibiting caspase-1 or IL-1β. These studies all indicated that inhibiting IL-1β was not a promising therapeutic target, which was consistent with human clinical trials[119]. However, these data do not necessarily disprove the role of pyroptosis in OA. First, the animal and cartilage explant models in these studies may not accurately reflect the aspects of the disease that are mediated by pyroptosis. Second, pyroptosis may be a process that is closely related to cell death, not just inflammatory factors such as IL-1β. ...
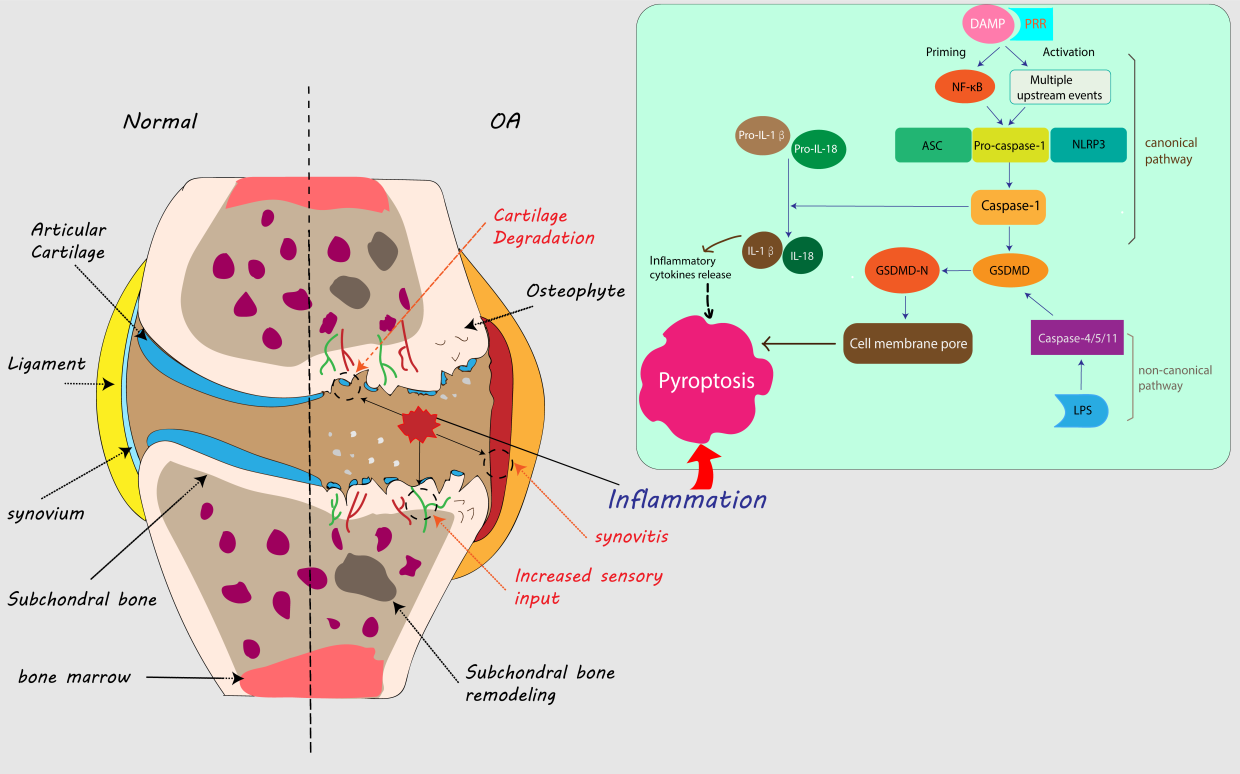
{kind=link}
{kind=link}



