Ding Yuejia1, Wang Yuan1, Zhang Wanqin1, Jia Qiujin1, Wang Xiaoling3, Li Yanyang4, Lv Shichao1, 2, *, Zhang Junping1, *
1First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, Tianjin 300193, China2Tianjin Key Laboratory of Traditional Research of TCM Prescription and Syndrome, Tianjin 300000, China3Qian’an Hospital of Traditional Chinese Medicine, Qian’an 064400, China4Tianjin Medical University Cancer Institute and Hospital, Tianjin 300060, China
通讯作者: Correspondence should be addressed to: Dr. Shichao Lv (email: 372272027@qq.com) and Junping Zhang (email: tjzhtcm@163.com), First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, Tianjin 300193, China.Correspondence should be addressed to: Dr. Shichao Lv (email: 372272027@qq.com) and Junping Zhang (email: tjzhtcm@163.com), First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, Tianjin 300193, China.Correspondence should be addressed to: Dr. Shichao Lv (email: 372272027@qq.com) and Junping Zhang (email: tjzhtcm@163.com), First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, Tianjin 300193, China.
收稿日期:2020-02-24
修回日期: 2020-06-3
接受日期: 2020-06-4
网络出版日期: 2020-10-01
版权声明:
2020 this is an open access article distributed under the terms of the creative commons attribution license, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
展开
Abstract
Myocardial fibrosis is observed in various cardiovascular diseases and plays a key role in the impairment of cardiac function. Endomyocardial biopsy, as the gold standard for the diagnosis of myocardial fibrosis, has limitations in terms of clinical application. Therefore, biomarkers have been recommended for noninvasive assessment of myocardial fibrosis. This review discusses the role of biomarkers in myocardial fibrosis from the perspective of collagen.
DingYuejia, WangYuan, ZhangWanqin, JiaQiujin, WangXiaoling, LiYanyang, LvShichao, ZhangJunping. Roles of Biomarkers in Myocardial Fibrosis[J]. Aging and Disease, 2020, 11(5): 1157-1174 https://doi.org/10.14336/AD.2020.0604
1.Introduction
Myocardial fibrosis is characterized by alterations of the extracellular matrix and is an integral part of most cardiac pathological conditions [1]. Currently, five types of collagen are known to be expressed in the myocardium. The cardiac extracellular matrix is primarily composed of fibrillar collagen type I (85%) and type III (11%) [2, 3]. Small amounts of collagen type IV and V are found in the basement membrane of myocytes and in the pericellular space [4, 5]. Besides, fibrillar collagen type VI is related to the adhesion of cellular fibers [6]. Extracellular collagen not only maintains cardiac fiber alignment, but also influences ventricle stiffness [7]. Moreover, an imbalance in collagen synthesis, breakdown, and metabolism accounts for the occurrence and development of myocardial fibrosis. Endomyocardial biopsy is undoubtedly the gold standard for the diagnosis of myocardial fibrosis, but it has limitations in clinical application. Therefore, biomarkers should be considered as a noninvasive method for detecting fibrosis. Myocardial fibrosis biomarkers have been investigated extensively, and fibrosis markers play an important role in the prognosis of cardiovascular diseases, such as heart failure (HF), dilated cardiomyopathy (DCM), and hypertrophic cardiomyopathy (HCM). This review summarizes the biomarkers of myocardial fibrosis from four collagen-related perspectives: synthesis, breakdown, metabolism, and gene transcription (miRNA).
2.Collagen Synthesis
2.1 C-Terminal Propeptide of Procollagen Type I
Type I collagen is a heterotrimeric molecule, which is composed of two α 1 chains and one α 2 chain. During its synthesis, the protein undergoes a series of post-translational modifications to form the procollagen chain; this precursor is then secreted into the extracellular space and cleaved by specific proteinases [8]. The stoichiometric ratio of the C-terminal propeptide of procollagen type I (PICP), which is released into the blood, and collagen type I produced by cleavage is 1:1 [9]. The heart secretes PICP into the peripheral circulation through the coronary sinus [9]. However, whether there is a positive correlation between plasma PICP, and myocardial collagen content remains controversial. A cross-sectional study demonstrated that plasma PICP levels in HCM patients were positively correlated with myocardial PICP content and the histological myocardial collagen volume fraction [10]. Similarly, Ferreira et al. also confirmed that the serum levels of PICP were significantly higher in patients with hypertension before drug treatment [11]. In contrast, in heart failure rats, Adamcova et al. observed that collagen content in the left ventricle was increased, whereas plasma PICP levels were reduced by 42% as compared to the control group [12]. These findings suggest that the myocardial collagen content does not necessarily correlate with plasma PICP, which may be related to confounding factors (such as weight loss and catabolic state). For predictive value, Ruiz-Ruiz et al. considered that serum PICP can be used independently for predicting HF episodes, hospital readmission, and death [13]. They included 111 patients with decompensated HF and found that 22.52% of the patients died during the 21 months of follow-up, and 48.6% of the patients were readmitted for HF. Moreover, serum PICP levels were significantly increased among patients who reached some primary endpoints (new hospitalizations or death) during follow-up (88.12 ± 37.31 ng/mL vs 73.13 ± 34.06 ng/mL; p = 0.029), and a cut-off value of 124 ng/mL predicted prognosis most accurately. Unfortunately, they did not perform cardiac biopsies, which is considered the most reliable method for measuring myocardial fibrosis.
2.2Procollagen Type I N-Terminal Propeptide
Excessive deposition of collagen type I is a feature of cardiac remodeling [14]. The procollagen type I N-terminal propeptide (PINP) originates from conversion of collagen type I, and is a marker for collagen type I synthesis [15]. N-terminal pro-peptides are removed by members of the ADAMTS family [16]. Compared with PICP, PINP has the disadvantage of delayed release [17]. At present, the relationship between PINP levels and fibrosis remains elusive. In a case?control study, Zile et al. observed that the baseline serum PINP levels in patients with HF were significantly higher than those in controls [18]. In rats with ischemic cardiomyopathy, Zhang et al. detected significant cardiac fibrosis by Masson staining 6 weeks after myocardial infarction (MI), and enzyme-linked immunosorbent assay (ELISA) results showed that the level of plasma PINP in the MI group was significantly increased as compared to that in the normal control group [19]. However, in a cross-sectional study in which serum PINP levels were measured by radioimmunoassay, no significant difference was revealed between HCM patients and healthy individuals [20]. As shown by the above studies, PINP has some limitations as a biomarker.
2.3. Procollagen Type III Amino-Terminal Propeptide
Collagen type III, whose fibers have a relatively small diameter, is synthesized by cardiac fibroblasts and is mainly responsible for myocardial elasticity [21, 22]. After the C-propeptides of fibrillar procollagen are cleaved by proteases, procollagen type III amino-terminal propeptide (PIIINP) is released into the blood via the lymphatics [17]. The N-propeptide is considered to play an important role in the regulation of primary fibril diameter [23]. Excellent storage and stability are the characteristics of PIIINP [24]. However, PIIINP is sometimes not completely separated from procollagen, which leads to the underestimation of type III collagen synthesis. In a prospective study of dilated cardiomyopathy, baseline serum PIIINP levels in patients with idiopathic or ischemic dilated cardiomyopathy were significantly higher than those in healthy controls, and serum PIIINP levels were highly correlated with cardiac collagen type III levels [25]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis.
3.Collagen Breakdown
3.1 C-Terminal Telopeptide of Collagen Type I
During degradation of collagen type I fibrils, the C-terminal telopeptide of collagen type I (CITP) is a cross-linked terminal peptide released in a 1:1 stoichiometric ratio, allowing accurate measurement of collagen degradation [30, 31]. In a cross-sectional study, serum CITP levels in HCM patients were increased, while the PICP and PINP levels were not altered significantly, indicating a shift in the collagen balance toward collagen type I breakdown [20]. This is an important outcome, because increased myocardial stiffness is usually caused by collagen deposition. However, several other studies have shown that the relationship between CITP and myocardial fibrosis is controversial. In patients with HF and atrial fibrillation (AF), serum CITP levels were significantly higher than those in the control groups [32, 33]. But Nagao et al. observed that serum CITP levels in patients with DCM were not associated with left ventricular remodeling parameters, or with the expression of cardiac collagen type I and type III [34]. In addition, some studies have confirmed the predictive value of CITP. Manhenke et al. considered that plasma CITP was an independent predictor of cardiovascular mortality in patients with acute myocardial infarction (AMI) [35]. This prospective study included 233 patients with AMI, among whom 56% reached the combined endpoint of HF symptoms or CV death during the years of follow-up, and plasma ICTP was increased in patients who died due to any cause. Similarly, serum CITP is also useful for determining cardiac events in patients with complete HF [36]. In summary, CITP may facilitate diagnosis or prognosis of myocardial fibrosis.
3.2 Matrix metalloproteinases
Matrix metalloproteinases (MMPs) are zinc- and calcium-dependent peptide enzymes that are involved in myocardial remodeling and degradation of collagen [37, 38]. It promotes the degradation of extracellular matrix, such as collagen, elastin, and gelatin [39]. MMPs can hydrolyze collagen efficiently, but their relative activities towards interstitial collagens are different. At 25°C, MMP-1 preferentially cleaves collagen type III over types I and II [40]. MMP-13 cleaves collagen type II five-fold faster than collagen type I and six-fold more rapidly than collagen type III, at 25 °C [41]. For MMP-2 and MMP-9, collagen type III is the substrate preferred over collagen types I and II [42, 43].
Many MMPs are expressed in the myocardium. In HCM, the expression of MMPs is altered, but not all MMPs are affected. Münch et al. found that MMP-1, MMP-2, MMP-3, and MMP-9 are involved in HCM [44]. They observed that increased serum MMP-2 levels in females were associated with lower fibrosis, while MMP-9 was positively associated with fibrosis in late gadolinium enhancement cardiac magnetic resonance (mean increase of 0.66 g/unit MMP-9 [0.50;0.82], p < 0.001), but neither serum MMP-1 nor MMP-3 levels were associated with cardiac fibrosis [44]. However, a cautionary note on these results is that the cardiac tissue concentration of MMPs was not evaluated, which might present a limitation. Regarding MMP-1 levels in systolic and diastolic HF, serum MMP-1 levels were increased in systolic HF patients, but not in diastolic HF patients [45, 46]. This result reminds us that MMP activity also fluctuates because heart remodeling during HF is a dynamic process. In diabetic mice (DM), myocardial fibrosis is clearly related to the expression of MMP7, MMP11, MMP13, and MMP16 in myocardial tissue [47]. Compared with the control group, levels of MMP7, MMP11, MMP13, and MMP16 were markedly higher, but levels of MMP2 were markedly lower in the DM group [47]. It is also important to note the measurement method of MMPs in all the above studies. The levels of different MMPs are determined by immunohistochemistry or histology or by enzyme-linked immunosorbent assays, but these methods do not distinguish proMMPs from active MMPs. Therefore, these results are of limited value.
3.3 Tissue Inhibitors of Metalloproteinase
Tissue inhibitors of metalloproteinase (TIMPs) are a group of low molecular weight glycoproteins. TIMPs are produced and secreted by fibroblasts, epithelial cells, and endothelial cells, and are distributed among tissues and humors [48]. TIMPs can promote the differentiation of fibroblasts into myofibroblasts at the site of tissue injury [49]. In addition, TIMPs control the proteolytic activity of MMPs [50]. Specifically, TIMPs can specifically bind to the zinc ions in the catalytic site of MMPs through the cysteine residues, thus disbanding activated MMPs, or preventing inactive MMPs from becoming activated [47]. Therefore, the balance between MMP and TIMP expression is essential for reconstruction of the extracellular matrix in myocardial tissues [51].
Currently, 4 types of TIMPs have been found to be associated with myocardial fibrosis. TIMP1 mediates the interaction between fibroblast membrane protein CD63 and integrin β1, which is widely expressed in various cells, thus triggering fibrosis [52-55]. At the cellular level, hirudin reverses Ang II-induced fibrosis by elevating TIMP-2 expression [56]. TIMP3 is the only TIMP that can inhibit the activity of TNF-α-converting enzyme and may have a protective effect against fibrosis by upregulating cytokines involved in myofibroblast activation and immunity [57]. In addition, Rizzi et al. investigated whether there was a correlation between cardiac TIMP-4 levels and cardiac hypertrophy in 2K1C hypertension rats [58]. They observed higher TIMP-4 levels (325 ± 68%) in hypertensive rats than in sham-operated animals at 75 days after 2K1C surgery. This result suggested that the increase in TIMP-4 activity was associated with concomitant development of cardiac hypertrophy. Another way to determine the severity of fibrosis is by measuring the MMP/TIMP ratio. Parkkonen et al. observed a large increase in serum TIMP-1 and MMP-2 levels in patients with takotsubo cardiomyopathy (TTC) compared with that in healthy controls, and proposed that the low MMP-8/TIMP-1 molar ratio may reflect increased transient fibrosis and decreased proteolysis [59]. This result indicates that the TIMP/MMP balance may play an important role in the development of myocardial fibrosis. In addition, the predicted value of TIMP-1 is also noteworthy. Frantz et al. investigated the association between TIMP-1 plasma levels and clinical endpoints (death due to any cause), and found that plasma TIMP-1 levels increased in HF patients as compared with healthy controls (1640 vs 735 ng/mL, respectively,) [60]. Moreover, the researchers also observed that patients with high TIMP-1 levels (1917 ng/mL) had a significantly higher mortality rate than patients with lower levels (1390 ng/mL). These results suggest that an increase in TIMP-1 indicates a poor prognosis in HF patients.
The collagen synthesis and degradation processes are briefly described in Fig. 1.
Figure 1. The process of collagen synthesis and degradation.
4.Collagen Metabolism
4.1 Transforming Growth Factor-β and Smads
Transforming growth factor-β (TGF-β) is a crucial profibrotic cytokine involved in myocardial fibrosis [61]. It is involved in the regulation of fibroblast proliferation, transformation, and migration, and production of the extracellular matrix [62]. As a cytokine upstream of LOX in cardiac fibroblasts, TGF-β activates fibrosis via the Smad3, PI3K/Akt, and MAPK signaling pathways [63]. In addition, the heterodimeric cell surface receptor complex composed of type I TGF-β receptors and type II TGF-β receptors plays an important role in signal transduction of cardiac fibroblasts [64, 65].
In various animal models of heart disease, inhibition of the effect of TGF-β successfully reduced or prevented the development of fibrosis. In MI mice, anti-TGF-β treatment after coronary artery ligation increased the expression of MMPs and decreased the production of collagen [66]. Similarly, in pressure-overloaded rats, the delivery of anti-TGF-β neutralizing antibody inhibited the activation of fibroblasts and prevented the induction of collagen type I and type III mRNA [67]. These observations indicated that anti-TGF treatment prevented excessive extracellular matrix deposition. Moreover, Koitabashi et al. observed that deletion of the TGF-β type II receptor (TβR2) gene in cardiomyocytes inhibited cardiac hypertrophy, and that transverse aortic constriction (TAC) mice receiving anti-TGF agents displayed significantly suppressed perivascular and interstitial fibrosis [68]. Taken together, these data emphasize the key role of TGF-β in cardiac fibrosis.
Smads, as key intracellular effectors of the TGF-β1 class, and play a central role in cardiac remodeling [69, 70]. Research has suggested that the up-regulatory effect of TGF-β3 on the migration, proliferation, and collagen synthesis of human cardiac fibroblasts may be attributed to the action of Smad7 [71]. Khalil et al. found that the absence of Smad3, Smad2/3, or Tgfbr1/2 markedly attenuated cardiac fibrosis after 12 weeks of TAC stimulation in rats [72]. In Smad3-deficient mice, the collagen content in the infarcted hearts decreased at 7 days after reperfusion, and collagen deposition in the uninfarcted, remodeling myocardium was markedly attenuated [73]. These findings suggest that Smad gene disruption leads to attenuation of the remodeling process.
4.2 Connective Tissue Growth Factor
Connective tissue growth factor (CTGF) belongs to the CCN family of multifunctional matricellular proteins, and it is involved in the process of triggering fibrosis in multiple organs and tissues, including the heart [74, 75]. Many in vitro studies have demonstrated that CTGF promotes the differentiation of fibroblasts into myofibroblasts and enhances extracellular matrix production [76-78]. CTGF is also a critical mediator of the downstream signaling of the profibrotic cytokine TGF-β [79, 80]. In DCM model rats, CTGF/CCN2 gene silencing improved cardiac function, attenuated myocardial fibrosis, and left ventricular hypertrophy [81]. In an in vitro study, β-adrenergic receptor overstimulation could induce the synthesis and secretion of CTGF in cardiomyocytes, thereby affecting the activation of cardiac fibroblasts [82]. Dean et al. observed the expression of spatiotemporal CTGF during the development of myocardial fibrosis in experimental MI rats [83]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis.
4.3 Corin
Corin is a transmembrane protease that is mainly expressed in cardiomyocytes [85]. It is closely correlated with natriuretic peptides that regulate many signaling functions, such as cGMP levels, vasodilation, natriuresis, fibrosis, etc. [86, 87]. In DCM model mice, increasing the expression of cardiac corin not only reduced fibrosis and HF, but also increased survival [88]. This finding indicates that corin correlates with the prognosis of DCM. In a cross-sectional study, plasma corin levels were lower in HF patients (365 pg/mL [±SD, 259 pg/mL]; P < 0.001) than in healthy controls, and this reduction was closely related to the severity of the disease (P < 0.001 for NYHA class II vs. class IV; P < 0.05 for NYHA class III vs. class IV) [85]. The results demonstrated that plasma corin may indicate pathological conditions in the heart. In canines with HF, the staining intensity of collagen was markedly elevated, while the expression of corin mRNA and protein was lower than that in the control group [89]. Contrary to these findings, Tran et al. observed upregulation of corin gene expression in the failing myocardium and in hypertrophic cardiomyocytes [90]. These studies suggest that the expression of corin may differ according to the disease state. In summary, corin may be an attractive diagnostic and prognostic biomarker of myocardial fibrosis.
4.4 Mesenchymal cell products
Recently, endothelial-to-mesenchymal transition (EndoMT) has been considered as a potential mechanism in pathological fibrosis. In the complex biological process of EndoMT, endothelial cells lose their specific cellular markers, and then acquire a mesenchymal or myofibroblast phenotype to initiate expression of mesenchymal cell products, such as α-smooth muscle actin (α-SMA), vimentin, and fibronectin [91]. In a rat model of isoproterenol (ISO) -induced myocardial fibrosis, cardiac α-SMA and vimentin levels increased, peaking on day 3, and then gradually decreased [92]. This result suggests that α-SMA and vimentin activity fluctuates in the fibrosis process. α-SMA, fibronectin, and vimentin are also involved in the process of diabetic cardiomyopathy [93]. The immunohistochemistry data showed that diabetes enhanced the expression of cardiac α-SMA, fibronectin, and vimentin compared with normal rats. Moreover, in vitro treatment of mouse embryonic fibroblasts with WF-A reduced the stability of collagen mRNA, but in the absence of vimentin, WF-A did not change the half-life of collagen mRNAs [94]. We therefore conclude that vimentin filaments play a crucial role in collagen expression. In short, α-SMA, vimentin, and fibronectin might be essential elements in cardiac fibrosis.
4.5 Inflammatory Factors
Inflammation caused by infection and tissue necrosis is a physiological response that promotes tissue healing through fibrosis, but it may become excessive when accompanied by additional factors, such as mechanical stress, genetic background, activation of neurohumoral factors, oxidative stress, and autoimmunity, causing pathological remodeling through Gal-3, TNF-α, interleukin, MMPs, miRNA activation, and other mechanisms.
Galectin-3 (Gal-3), a β-galactoside-binding lectin, plays a regulatory role in fibrogenesis. It is widely expressed by various types of inflammatory cells, binds to extracellular glycoproteins, and activates fibroblasts to increase collagen I deposition [95-97]. Growing evidence indicates that high levels of circulating Gal-3 are associated with an increased risk of adverse cardiovascular events, such as HF, myocardial infarction, dilated cardiomyopathy, and fibrogenesis [98-101]. In mice with ischemia/reperfusion (I/R) injury, there was a two-fold increase in plasma Gal-3 levels, and in the ischemic myocardium, Gal-3 was upregulated seven-fold at the mRNA level and 30-fold at the protein level [102]. In addition, that study revealed that plasma Gal-3 concentrations were always higher in DCM/ICM patients with HF than in non-HF subjects, but no trans-cardiac or trans-hepatic concentration gradient of Gal-3 was found. The aforementioned studies were limited in that they did not obtain myocardial biopsies to measure cardiac Gal-3 expression. Moreover, serum Gal-3 is considered to be associated with inflammation and cardiovascular fibrosis. In patients with AMI, serum Gal-3 levels increased immediately after AMI and then declined significantly within 5 days [103]. These data indicate that Gal-3 is closely related to the formation, destabilization, and rupture of plaque. Besides observational studies of Gal-3 levels, several studies have investigated the predictive value of Gal-3. Van Kimmenade et al. observed that an increased circulating Gal-3 level was the best independent predictor of 60-day mortality (odds ratio 10.3, P < 0.01) in patients with acute HF [104]. In patients with coronary artery disease (CAD), Maiolino et al. found that patients with Gal-3 levels in the highest tertile were more prone to death due to cardiovascular causes (25.2%) than those with Gal-3 levels in the intermediate and lower tertiles (13.6% and 7.5%, respectively; P < 0.001), and the plasma Gal-3 cutoff value for cardiovascular death prediction was 27.7 ng/mL [105]. Unfortunately, the loss of 25% of the patients to follow-up may have limited the results of the study. Overall, these findings confirm that elevated Gal-3 is associated with cardiovascular disease progression and poor outcome.
A growing body of evidence has demonstrated that macrophages and monocytes not only play a key role in the occurrence and development of fibrotic responses, but may also mediate the regression of fibrosis [106]. Macrophages and monocytes are capable of producing and excreting a large number of pro-inflammatory mediators, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-4, and IL-6 [107]. Furthermore, a complex regulatory network consisting of these inflammatory factors activates both myofibroblasts and fibrotic pathological processes [107]. This was supported by Kang et al., who treated primary cardiomyocytes with 35 mmol/L glucose for 24 h [108]. The ELISA results showed that the levels of TNF-α, IL-6, and IL-1β and the expression of collagen I and III mRNA were higher than those in the normal control group [108]. These findings suggest that fibrosis is associated with inflammation. In a cross-sectional study of HCM, Fang et al. observed that plasma IL-4, IL-6, and IL-10 levels were positively correlated with diffuse and regional myocardial fibrosis [97]. Unfortunately, owing to the cross-sectional nature of the research, it is impossible to determine whether this association was causal. In addition, in order to study the role of IL-11 signal transduction in cardiovascular fibrosis, Schafer et al. generated IL-11 knockout mice. Compared with wild-type mice, they observed less cardiac fibrosis in knockout mice after either transverse aortic constriction or AngII infusion, suggesting that IL11 is a crucial profibrotic gene [109]. In summary, inflammatory factors are involved in the pathological process of myocardial fibrosis.
Table 1
Table 1 Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
MMPs and miRNAs also play a regulatory role in the process of inflammatory fibrosis. Tissue injury induces activation of inflammatory mediators and recruitment of inflammatory cells. Inflammatory cells promote fibrosis by producing cytokines/chemokines that regulate MMPs [110]. MMP-9, the most widely documented protease in the inflammatory process, is involved in the initial stage of myocardial injury. The increase in MMP-9 levels is related to the invasion of necrotic tissue by inflammatory cells, particularly polymorphonuclear neutrophils and activated satellite cells [111]. Moreover, in the cardiovascular system, miRNAs control the expression of some inflammatory factors. For example, miR-155 controls the expression of SHIP1 and SOCS1, the key regulators of the inflammatory response in macrophages [112]. Elevated miR-21 expression in macrophages inhibited the production of TNF-α and the up-regulation of IL-10, respectively [113]. Overall, these inflammatory markers are associated with fibrosis.
3.6Other Molecules
The development of myocardial fibrosis also involves factors such as relaxin-1 (RLN1), growth hormone secretagogue receptor (GSHR), ADAMTS-1, and transient receptor potential melastatin 7 (TRPM7). RLN, a polypeptide hormone, is secreted by cardiac cells [114]. In a cross-sectional study, the average circulating RLN1 levels in the HFrEF patient population were markedly higher than those in healthy control subjects (702 ± 283 pg/mL vs. 44 ± 27 pg/mL), with elevated RLN1 levels accompanied by a decrease in heart fibrosis [115]. This finding promotes the study of RLN1 as a potential anti-fibrosis target. GSHR is an orexigenic hormone with newly defined cardiovascular effects. In a mouse model of isoproterenol-induced myocardial fibrosis, GHSR deficiency exacerbated the expression of myofibroblast trans-differentiation marker genes, suggesting that GHSR may be a surrogate indicator of the need for intervention in myocardial fibrosis [116]. ADAMTS-1, a metalloprotease with proteolytic activity, also has the ability to cleave the N-terminal propeptide of collagen. In mice with viral heart disease (VHD), Li et al. observed that the collagen volume fraction increased significantly, accompanied by elevated expression of ADAMTS-1 mRNA in cardiac tissue (P < 0.001) [117]. Moreover, TRPM7 is capable of promoting the proliferation and differentiation of fibroblasts and increasing the synthesis of extracellular matrix proteins [118].
In general, these molecules, which are closely related to collagen synthesis, breakdown, and metabolism, have recently been proposed as intervention targets for myocardial fibrosis (Table 1).
5.Gene Transcription (miRNA)
MicroRNAs (miRNAs) are small, noncoding RNAs that regulate gene expression after transcription, and play a critical role in heart function and pathology [119]. The miRNA family regulates gene expression by inducing target mRNA destabilization or inhibiting protein translation [120]. The marked cardiospecific capacity of some miRNAs is beneficial for ameliorating tissue remodeling.
The relationship between miR-21 and myocardial fibrosis has been extensively studied. miRNAs target not only single genes, but also overall networks that contribute to biological function. For example, in patients with aortic stenosis (AS), myocardial and plasmatic miR-21 levels predicted collagen type I, collagen type III, and fibronectin expression by targeting RECK, PDCD4, and TGF-β-signaling factors [121]. In animal models of myocardial infarction, miR-21 promoted the transformation of cardiac fibroblasts (CFs) to myofibroblasts, and increased myocardial fibrosis in vivo by targeting Jagged1 [122]. In addition, the overexpression of miR-21 in allogeneic mice activated the fibrosis gene program and promoted the differentiation of monocytes to fibroblasts via the phosphatase and tensin homologue/activator protein 1 regulatory axis (PTEN/AP-1) [123]. Intriguingly, Szemraj-Rogucka et al., in a study of 13 left ventricular non-compaction patients (LVNC), found that plasma levels of miR-21, miR-29a, miR-30d, and miR-133a were both significantly elevated, suggesting that all four miRNAs may serve as biomarkers of myocardial fibrosis [124]. Unfortunately, this study was performed using a small sample size, which may have biased the results.
Table 2
Table 2 Biomarkers of myocardial fibrosis (gene transcription).
Figure 2. Biomarkers and pathological effects of myocardial fibrosis.
Another well-researched miRNA is miR-29, which demonstrates reduced expression under cardiac stress conditions, and thus may promote more extracellular matrix protein production by “derepression” of elastin, genes encoding collagens, and fibrillin [125]. Heid et al. found that overexpression of the miR-29 family counteracted the physiological accumulation of oxidative damage during aging, thereby protecting the heart against the detrimental effects of fibrosis [126]. In patients with pathological hypertrophy, TaqMan quantitative polymerase chain reaction showed a marked decrease in cardiac miR-29a and miR-29c, suggesting that miR-29 expression has been linked to extracellular matrix remodeling in cardiac hypertrophy [127]. MiR-29 is also a good example of a potential therapeutic target. For example, infusion of anti-miR-29 into cardiac hypertrophy model mice could prevent the expression of fibrosis markers, such as Col1a1, Col1a2, and Col3a1 [128]. Together, the miR-29 family plays a key role in pathological hypertrophy of the myocardium and fibrosis.
Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2).
6.Conclusion
Myocardial fibrosis, as a main component of most cardiovascular diseases, has been a major focus in recent years. Endomyocardial biopsy, which is the gold standard for the diagnosis of myocardial fibrosis, has limitations in terms of clinical application, whereas biomarkers seem to be easier and safer in terms of diagnosis, therapeutic monitoring, and prognosis. With the development of technology, the investigation of myocardial fibrosis biomarkers has received attention in clinical and research communities. A systematic review of biomarkers and pathological effects of myocardial fibrosis is presented in Fig. 2.
When selecting biomarkers in experiments, researchers should consider the purpose and method of the experiment. Some biomarkers are very likely “bystander” markers, but many are functional factors that are closely related to collagen synthesis and degradation. Specifically, PICP, PINP, and PIIINP are suitable representatives of the mechanism of collagen synthesis in target organ injury in myocardial fibrosis. CITP, MMPs, and TIMPs reflect collagen degradation, and the balance of collagen synthesis and degradation in turn indicates the stability of organ fibrosis. Therefore, PICP, PINP, PIIINP, CITP, MMPs, and TIMPs are functional factors that can directly reflect the degree of fibrosis. Moreover, in the process of fibrosis, collagen metabolism is affected by many molecules, such as TGF-β, Smads, CTGF, corin, mesenchymal cell products, and inflammatory factors. CTGF induces proliferation of fibroblasts and increases extracellular matrix content. Corin affects heart function by regulating natriuretic peptides. Inflammation always accompanies fibrosis, and hence inflammatory markers can reflect the relationship between them. EndoMT is one of the important sources of fibroblasts, and TGF-β, Smads, and miRNA are the main regulators of collagen gene expression. Therefore, TGF-β, Smads, CTGF, corin, mesenchymal cell products, and inflammatory factors are “bystander” markers that can indirectly affect the fibrosis process.
It is important to note that fibrosis occurs not only in the heart, but also in other organs, so that changes in biomarker levels may not have only a cardiac origin [25]. In another respect, biomarkers must be strictly tested to determine whether they strongly reflect myocardial fibrosis. Endomyocardial biopsy can be used to estimate the usefulness and accuracy of biomarkers. Only when these initiatives are successful can biomarkers be incorporated into clinical practice. Additionally, cost-effectivity issues should also be taken into consideration, as the measurement of many biomarkers mentioned is not cheap, particularly when using a multiple-biomarker approach. Moreover, there is not currently a well-tested biomarker for fibrosis that is equivalent to NT-proBNP for HF. On the whole, the use of biomarkers is helpful in the assessment of myocardial fibrosis; therefore, more prospective studies are needed in the future.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81904118 and 81603559) and Young Elite Scientists Sponsorship Program by CAST (No. CACM-2018-QNRC2-B04).
YangC, QiaoS, SongY, LiuY, TangY, DengL, et al. (2019).
Procollagen type I carboxy-terminal propeptide (PICP) and MMP-2 are potential biomarkers of myocardial fibrosis in patients with hypertrophic cardiomyopathy
GuhaN, Erotokritou-MulliganI, BartlettC, CowanDA, BassettEE, StowM, et al. (2012).
The effects of a freeze-thaw cycle and pre-analytical storage temperature on the stability of insulin-like growth factor-I and pro-collagen type III N-terminal propeptide concentrations: Implications for the detection of growth hormone misuse in athletes
KlappacherG, FranzenP, HaabD, MehrabiM, BinderM, PleschK, et al. (1995).
Measuring extracellular matrix turnover in the serum of patients with idiopathic or ischemic dilated cardiomyopathy and impact on diagnosis and prognosis
LinYT, LinYH, WuXM, KoCL, YenRF, ChenYH, et al. (2017).
The relationship between serum fibrosis markers and restrictive ventricular filling in patients with heart failure with reduced ejection fraction: A technetium-99m radionuclide ventriculography study
Serum concentration of procollagen type III amino-terminal peptide is increased in patients with successfully repaired coarctation of the aorta with left ventricular hypertrophy
KitaharaT, TakeishiY, ArimotoT, NiizekiT, KoyamaY, SasakiT, et al. (2007).
Serum carboxy-terminal telopeptide of type I collagen (ICTP) predicts cardiac events in chronic heart failure patients with preserved left ventricular systolic function
MünchJ, AvanesovM, BannasP, SäringD, KrämerE, MeariniG, et al. (2016).
Serum Matrix Metalloproteinases as Quantitative Biomarkers for Myocardial Fibrosis and Sudden Cardiac Death Risk Stratification in Patients With Hypertrophic Cardiomyopathy
ParkkonenO, NieminenMT, VesterinenP, TervahartialaT, PerolaM, SalomaaV, et al. (2017).
Low MMP-8/TIMP-1 reflects left ventricle impairment in takotsubo cardiomyopathy and high TIMP-1 may help to differentiate it from acute coronary syndrome
TGF-β1/Smad signaling pathway regulates epithelial-to-mesenchymal transition in esophageal squamous cell carcinoma: in vitro and clinical analyses of cell lines and nomadic Kazakh patients from northwest Xinjiang, China
DanielsJT, SchultzGS, BlalockTD, GarrettQ, GrotendorstGR, DeanNM, et al. (2003).
Mediation of transforming growth factor-beta(1)-stimulated matrix contraction by fibroblasts: a role for connective tissue growth factor in contractile scarring
Silencing CTGF/CCN2 inactivates the MAPK signaling pathway to alleviate myocardial fibrosis and left ventricular hypertrophy in rats with dilated cardiomyopathy
Systemic inflammation is associated with myocardial fibrosis, diastolic dysfunction, and cardiac hypertrophy in patients with hypertrophic cardiomyopathy
Up-regulation of microRNA-203 inhibits myocardial fibrosis and oxidative stress in mice with diabetic cardiomyopathy through the inhibition of PI3K/Akt signaling pathway via PIK3CA
Overexpression of microRNA-202-3p protects against myocardial ischemia-reperfusion injury through activation of TGF-β1/Smads signaling pathway by targeting TRPM6
RubiśP, Totoń-ŻurańskaJ, Wiśniowska-ŚmiałekS, HolcmanK, Kołton-WróżM, WołkowP, et al. (2017).
Relations between circulating microRNAs (miR-21, miR-26, miR-29, miR-30 and miR-133a), extracellular matrix fibrosis and serum markers of fibrosis in dilated cardiomyopathy
LiuW, ZhengJ, DongJ, BaiR, SongD, MaX, et al. (2018).
Association of miR-197-5p, a Circulating Biomarker for Heart Failure, with Myocardial Fibrosis and Adverse Cardiovascular Events among Patients with Stage C or D Heart Failure
PanZ, SunX, ShanH, WangN, WangJ, RenJ, et al. (2012).
MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-β1 pathway
... Myocardial fibrosis is characterized by alterations of the extracellular matrix and is an integral part of most cardiac pathological conditions [1]. Currently, five types of collagen are known to be expressed in the myocardium. The cardiac extracellular matrix is primarily composed of fibrillar collagen type I (85%) and type III (11%) [2, 3]. Small amounts of collagen type IV and V are found in the basement membrane of myocytes and in the pericellular space [4, 5]. Besides, fibrillar collagen type VI is related to the adhesion of cellular fibers [6]. Extracellular collagen not only maintains cardiac fiber alignment, but also influences ventricle stiffness [7]. Moreover, an imbalance in collagen synthesis, breakdown, and metabolism accounts for the occurrence and development of myocardial fibrosis. Endomyocardial biopsy is undoubtedly the gold standard for the diagnosis of myocardial fibrosis, but it has limitations in clinical application. Therefore, biomarkers should be considered as a noninvasive method for detecting fibrosis. Myocardial fibrosis biomarkers have been investigated extensively, and fibrosis markers play an important role in the prognosis of cardiovascular diseases, such as heart failure (HF), dilated cardiomyopathy (DCM), and hypertrophic cardiomyopathy (HCM). This review summarizes the biomarkers of myocardial fibrosis from four collagen-related perspectives: synthesis, breakdown, metabolism, and gene transcription (miRNA). ...
Collagen remodeling of the pressure-overloaded, hypertrophied nonhuman primate myocardium
1
1988
... Myocardial fibrosis is characterized by alterations of the extracellular matrix and is an integral part of most cardiac pathological conditions [1]. Currently, five types of collagen are known to be expressed in the myocardium. The cardiac extracellular matrix is primarily composed of fibrillar collagen type I (85%) and type III (11%) [2, 3]. Small amounts of collagen type IV and V are found in the basement membrane of myocytes and in the pericellular space [4, 5]. Besides, fibrillar collagen type VI is related to the adhesion of cellular fibers [6]. Extracellular collagen not only maintains cardiac fiber alignment, but also influences ventricle stiffness [7]. Moreover, an imbalance in collagen synthesis, breakdown, and metabolism accounts for the occurrence and development of myocardial fibrosis. Endomyocardial biopsy is undoubtedly the gold standard for the diagnosis of myocardial fibrosis, but it has limitations in clinical application. Therefore, biomarkers should be considered as a noninvasive method for detecting fibrosis. Myocardial fibrosis biomarkers have been investigated extensively, and fibrosis markers play an important role in the prognosis of cardiovascular diseases, such as heart failure (HF), dilated cardiomyopathy (DCM), and hypertrophic cardiomyopathy (HCM). This review summarizes the biomarkers of myocardial fibrosis from four collagen-related perspectives: synthesis, breakdown, metabolism, and gene transcription (miRNA). ...
Characterisation of left ventricular collagen in the rat
1
1983
... Myocardial fibrosis is characterized by alterations of the extracellular matrix and is an integral part of most cardiac pathological conditions [1]. Currently, five types of collagen are known to be expressed in the myocardium. The cardiac extracellular matrix is primarily composed of fibrillar collagen type I (85%) and type III (11%) [2, 3]. Small amounts of collagen type IV and V are found in the basement membrane of myocytes and in the pericellular space [4, 5]. Besides, fibrillar collagen type VI is related to the adhesion of cellular fibers [6]. Extracellular collagen not only maintains cardiac fiber alignment, but also influences ventricle stiffness [7]. Moreover, an imbalance in collagen synthesis, breakdown, and metabolism accounts for the occurrence and development of myocardial fibrosis. Endomyocardial biopsy is undoubtedly the gold standard for the diagnosis of myocardial fibrosis, but it has limitations in clinical application. Therefore, biomarkers should be considered as a noninvasive method for detecting fibrosis. Myocardial fibrosis biomarkers have been investigated extensively, and fibrosis markers play an important role in the prognosis of cardiovascular diseases, such as heart failure (HF), dilated cardiomyopathy (DCM), and hypertrophic cardiomyopathy (HCM). This review summarizes the biomarkers of myocardial fibrosis from four collagen-related perspectives: synthesis, breakdown, metabolism, and gene transcription (miRNA). ...
Collagen chain mRNAs in isolated heart cells from young and adult rats
1
1988
... Myocardial fibrosis is characterized by alterations of the extracellular matrix and is an integral part of most cardiac pathological conditions [1]. Currently, five types of collagen are known to be expressed in the myocardium. The cardiac extracellular matrix is primarily composed of fibrillar collagen type I (85%) and type III (11%) [2, 3]. Small amounts of collagen type IV and V are found in the basement membrane of myocytes and in the pericellular space [4, 5]. Besides, fibrillar collagen type VI is related to the adhesion of cellular fibers [6]. Extracellular collagen not only maintains cardiac fiber alignment, but also influences ventricle stiffness [7]. Moreover, an imbalance in collagen synthesis, breakdown, and metabolism accounts for the occurrence and development of myocardial fibrosis. Endomyocardial biopsy is undoubtedly the gold standard for the diagnosis of myocardial fibrosis, but it has limitations in clinical application. Therefore, biomarkers should be considered as a noninvasive method for detecting fibrosis. Myocardial fibrosis biomarkers have been investigated extensively, and fibrosis markers play an important role in the prognosis of cardiovascular diseases, such as heart failure (HF), dilated cardiomyopathy (DCM), and hypertrophic cardiomyopathy (HCM). This review summarizes the biomarkers of myocardial fibrosis from four collagen-related perspectives: synthesis, breakdown, metabolism, and gene transcription (miRNA). ...
Localization of types I, III and IV collagen mRNAs in rat heart cells by in situ hybridization
1
1989
... Myocardial fibrosis is characterized by alterations of the extracellular matrix and is an integral part of most cardiac pathological conditions [1]. Currently, five types of collagen are known to be expressed in the myocardium. The cardiac extracellular matrix is primarily composed of fibrillar collagen type I (85%) and type III (11%) [2, 3]. Small amounts of collagen type IV and V are found in the basement membrane of myocytes and in the pericellular space [4, 5]. Besides, fibrillar collagen type VI is related to the adhesion of cellular fibers [6]. Extracellular collagen not only maintains cardiac fiber alignment, but also influences ventricle stiffness [7]. Moreover, an imbalance in collagen synthesis, breakdown, and metabolism accounts for the occurrence and development of myocardial fibrosis. Endomyocardial biopsy is undoubtedly the gold standard for the diagnosis of myocardial fibrosis, but it has limitations in clinical application. Therefore, biomarkers should be considered as a noninvasive method for detecting fibrosis. Myocardial fibrosis biomarkers have been investigated extensively, and fibrosis markers play an important role in the prognosis of cardiovascular diseases, such as heart failure (HF), dilated cardiomyopathy (DCM), and hypertrophic cardiomyopathy (HCM). This review summarizes the biomarkers of myocardial fibrosis from four collagen-related perspectives: synthesis, breakdown, metabolism, and gene transcription (miRNA). ...
Molecular assembly, secretion, and matrix deposition of type VI collagen
1
1986
... Myocardial fibrosis is characterized by alterations of the extracellular matrix and is an integral part of most cardiac pathological conditions [1]. Currently, five types of collagen are known to be expressed in the myocardium. The cardiac extracellular matrix is primarily composed of fibrillar collagen type I (85%) and type III (11%) [2, 3]. Small amounts of collagen type IV and V are found in the basement membrane of myocytes and in the pericellular space [4, 5]. Besides, fibrillar collagen type VI is related to the adhesion of cellular fibers [6]. Extracellular collagen not only maintains cardiac fiber alignment, but also influences ventricle stiffness [7]. Moreover, an imbalance in collagen synthesis, breakdown, and metabolism accounts for the occurrence and development of myocardial fibrosis. Endomyocardial biopsy is undoubtedly the gold standard for the diagnosis of myocardial fibrosis, but it has limitations in clinical application. Therefore, biomarkers should be considered as a noninvasive method for detecting fibrosis. Myocardial fibrosis biomarkers have been investigated extensively, and fibrosis markers play an important role in the prognosis of cardiovascular diseases, such as heart failure (HF), dilated cardiomyopathy (DCM), and hypertrophic cardiomyopathy (HCM). This review summarizes the biomarkers of myocardial fibrosis from four collagen-related perspectives: synthesis, breakdown, metabolism, and gene transcription (miRNA). ...
Structure and function of myocardial fibrillar collagen
1
1997
... Myocardial fibrosis is characterized by alterations of the extracellular matrix and is an integral part of most cardiac pathological conditions [1]. Currently, five types of collagen are known to be expressed in the myocardium. The cardiac extracellular matrix is primarily composed of fibrillar collagen type I (85%) and type III (11%) [2, 3]. Small amounts of collagen type IV and V are found in the basement membrane of myocytes and in the pericellular space [4, 5]. Besides, fibrillar collagen type VI is related to the adhesion of cellular fibers [6]. Extracellular collagen not only maintains cardiac fiber alignment, but also influences ventricle stiffness [7]. Moreover, an imbalance in collagen synthesis, breakdown, and metabolism accounts for the occurrence and development of myocardial fibrosis. Endomyocardial biopsy is undoubtedly the gold standard for the diagnosis of myocardial fibrosis, but it has limitations in clinical application. Therefore, biomarkers should be considered as a noninvasive method for detecting fibrosis. Myocardial fibrosis biomarkers have been investigated extensively, and fibrosis markers play an important role in the prognosis of cardiovascular diseases, such as heart failure (HF), dilated cardiomyopathy (DCM), and hypertrophic cardiomyopathy (HCM). This review summarizes the biomarkers of myocardial fibrosis from four collagen-related perspectives: synthesis, breakdown, metabolism, and gene transcription (miRNA). ...
Collagen cross-linking: insights on the evolution of metazoan extracellular matrix
1
2016
... Type I collagen is a heterotrimeric molecule, which is composed of two α 1 chains and one α 2 chain. During its synthesis, the protein undergoes a series of post-translational modifications to form the procollagen chain; this precursor is then secreted into the extracellular space and cleaved by specific proteinases [8]. The stoichiometric ratio of the C-terminal propeptide of procollagen type I (PICP), which is released into the blood, and collagen type I produced by cleavage is 1:1 [9]. The heart secretes PICP into the peripheral circulation through the coronary sinus [9]. However, whether there is a positive correlation between plasma PICP, and myocardial collagen content remains controversial. A cross-sectional study demonstrated that plasma PICP levels in HCM patients were positively correlated with myocardial PICP content and the histological myocardial collagen volume fraction [10]. Similarly, Ferreira et al. also confirmed that the serum levels of PICP were significantly higher in patients with hypertension before drug treatment [11]. In contrast, in heart failure rats, Adamcova et al. observed that collagen content in the left ventricle was increased, whereas plasma PICP levels were reduced by 42% as compared to the control group [12]. These findings suggest that the myocardial collagen content does not necessarily correlate with plasma PICP, which may be related to confounding factors (such as weight loss and catabolic state). For predictive value, Ruiz-Ruiz et al. considered that serum PICP can be used independently for predicting HF episodes, hospital readmission, and death [13]. They included 111 patients with decompensated HF and found that 22.52% of the patients died during the 21 months of follow-up, and 48.6% of the patients were readmitted for HF. Moreover, serum PICP levels were significantly increased among patients who reached some primary endpoints (new hospitalizations or death) during follow-up (88.12 ± 37.31 ng/mL vs 73.13 ± 34.06 ng/mL; p = 0.029), and a cut-off value of 124 ng/mL predicted prognosis most accurately. Unfortunately, they did not perform cardiac biopsies, which is considered the most reliable method for measuring myocardial fibrosis. ...
Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis
2
2004
... Type I collagen is a heterotrimeric molecule, which is composed of two α 1 chains and one α 2 chain. During its synthesis, the protein undergoes a series of post-translational modifications to form the procollagen chain; this precursor is then secreted into the extracellular space and cleaved by specific proteinases [8]. The stoichiometric ratio of the C-terminal propeptide of procollagen type I (PICP), which is released into the blood, and collagen type I produced by cleavage is 1:1 [9]. The heart secretes PICP into the peripheral circulation through the coronary sinus [9]. However, whether there is a positive correlation between plasma PICP, and myocardial collagen content remains controversial. A cross-sectional study demonstrated that plasma PICP levels in HCM patients were positively correlated with myocardial PICP content and the histological myocardial collagen volume fraction [10]. Similarly, Ferreira et al. also confirmed that the serum levels of PICP were significantly higher in patients with hypertension before drug treatment [11]. In contrast, in heart failure rats, Adamcova et al. observed that collagen content in the left ventricle was increased, whereas plasma PICP levels were reduced by 42% as compared to the control group [12]. These findings suggest that the myocardial collagen content does not necessarily correlate with plasma PICP, which may be related to confounding factors (such as weight loss and catabolic state). For predictive value, Ruiz-Ruiz et al. considered that serum PICP can be used independently for predicting HF episodes, hospital readmission, and death [13]. They included 111 patients with decompensated HF and found that 22.52% of the patients died during the 21 months of follow-up, and 48.6% of the patients were readmitted for HF. Moreover, serum PICP levels were significantly increased among patients who reached some primary endpoints (new hospitalizations or death) during follow-up (88.12 ± 37.31 ng/mL vs 73.13 ± 34.06 ng/mL; p = 0.029), and a cut-off value of 124 ng/mL predicted prognosis most accurately. Unfortunately, they did not perform cardiac biopsies, which is considered the most reliable method for measuring myocardial fibrosis. ...
... ]. The heart secretes PICP into the peripheral circulation through the coronary sinus [9]. However, whether there is a positive correlation between plasma PICP, and myocardial collagen content remains controversial. A cross-sectional study demonstrated that plasma PICP levels in HCM patients were positively correlated with myocardial PICP content and the histological myocardial collagen volume fraction [10]. Similarly, Ferreira et al. also confirmed that the serum levels of PICP were significantly higher in patients with hypertension before drug treatment [11]. In contrast, in heart failure rats, Adamcova et al. observed that collagen content in the left ventricle was increased, whereas plasma PICP levels were reduced by 42% as compared to the control group [12]. These findings suggest that the myocardial collagen content does not necessarily correlate with plasma PICP, which may be related to confounding factors (such as weight loss and catabolic state). For predictive value, Ruiz-Ruiz et al. considered that serum PICP can be used independently for predicting HF episodes, hospital readmission, and death [13]. They included 111 patients with decompensated HF and found that 22.52% of the patients died during the 21 months of follow-up, and 48.6% of the patients were readmitted for HF. Moreover, serum PICP levels were significantly increased among patients who reached some primary endpoints (new hospitalizations or death) during follow-up (88.12 ± 37.31 ng/mL vs 73.13 ± 34.06 ng/mL; p = 0.029), and a cut-off value of 124 ng/mL predicted prognosis most accurately. Unfortunately, they did not perform cardiac biopsies, which is considered the most reliable method for measuring myocardial fibrosis. ...
Procollagen type I carboxy-terminal propeptide (PICP) and MMP-2 are potential biomarkers of myocardial fibrosis in patients with hypertrophic cardiomyopathy
2
2019
... Type I collagen is a heterotrimeric molecule, which is composed of two α 1 chains and one α 2 chain. During its synthesis, the protein undergoes a series of post-translational modifications to form the procollagen chain; this precursor is then secreted into the extracellular space and cleaved by specific proteinases [8]. The stoichiometric ratio of the C-terminal propeptide of procollagen type I (PICP), which is released into the blood, and collagen type I produced by cleavage is 1:1 [9]. The heart secretes PICP into the peripheral circulation through the coronary sinus [9]. However, whether there is a positive correlation between plasma PICP, and myocardial collagen content remains controversial. A cross-sectional study demonstrated that plasma PICP levels in HCM patients were positively correlated with myocardial PICP content and the histological myocardial collagen volume fraction [10]. Similarly, Ferreira et al. also confirmed that the serum levels of PICP were significantly higher in patients with hypertension before drug treatment [11]. In contrast, in heart failure rats, Adamcova et al. observed that collagen content in the left ventricle was increased, whereas plasma PICP levels were reduced by 42% as compared to the control group [12]. These findings suggest that the myocardial collagen content does not necessarily correlate with plasma PICP, which may be related to confounding factors (such as weight loss and catabolic state). For predictive value, Ruiz-Ruiz et al. considered that serum PICP can be used independently for predicting HF episodes, hospital readmission, and death [13]. They included 111 patients with decompensated HF and found that 22.52% of the patients died during the 21 months of follow-up, and 48.6% of the patients were readmitted for HF. Moreover, serum PICP levels were significantly increased among patients who reached some primary endpoints (new hospitalizations or death) during follow-up (88.12 ± 37.31 ng/mL vs 73.13 ± 34.06 ng/mL; p = 0.029), and a cut-off value of 124 ng/mL predicted prognosis most accurately. Unfortunately, they did not perform cardiac biopsies, which is considered the most reliable method for measuring myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Potential spironolactone effects on collagen metabolism biomarkers in patients with uncontrolled blood pressure
2
2019
... Type I collagen is a heterotrimeric molecule, which is composed of two α 1 chains and one α 2 chain. During its synthesis, the protein undergoes a series of post-translational modifications to form the procollagen chain; this precursor is then secreted into the extracellular space and cleaved by specific proteinases [8]. The stoichiometric ratio of the C-terminal propeptide of procollagen type I (PICP), which is released into the blood, and collagen type I produced by cleavage is 1:1 [9]. The heart secretes PICP into the peripheral circulation through the coronary sinus [9]. However, whether there is a positive correlation between plasma PICP, and myocardial collagen content remains controversial. A cross-sectional study demonstrated that plasma PICP levels in HCM patients were positively correlated with myocardial PICP content and the histological myocardial collagen volume fraction [10]. Similarly, Ferreira et al. also confirmed that the serum levels of PICP were significantly higher in patients with hypertension before drug treatment [11]. In contrast, in heart failure rats, Adamcova et al. observed that collagen content in the left ventricle was increased, whereas plasma PICP levels were reduced by 42% as compared to the control group [12]. These findings suggest that the myocardial collagen content does not necessarily correlate with plasma PICP, which may be related to confounding factors (such as weight loss and catabolic state). For predictive value, Ruiz-Ruiz et al. considered that serum PICP can be used independently for predicting HF episodes, hospital readmission, and death [13]. They included 111 patients with decompensated HF and found that 22.52% of the patients died during the 21 months of follow-up, and 48.6% of the patients were readmitted for HF. Moreover, serum PICP levels were significantly increased among patients who reached some primary endpoints (new hospitalizations or death) during follow-up (88.12 ± 37.31 ng/mL vs 73.13 ± 34.06 ng/mL; p = 0.029), and a cut-off value of 124 ng/mL predicted prognosis most accurately. Unfortunately, they did not perform cardiac biopsies, which is considered the most reliable method for measuring myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Relations between markers of cardiac remodelling and left ventricular collagen in an isoproterenol-induced heart damage model
2
2019
... Type I collagen is a heterotrimeric molecule, which is composed of two α 1 chains and one α 2 chain. During its synthesis, the protein undergoes a series of post-translational modifications to form the procollagen chain; this precursor is then secreted into the extracellular space and cleaved by specific proteinases [8]. The stoichiometric ratio of the C-terminal propeptide of procollagen type I (PICP), which is released into the blood, and collagen type I produced by cleavage is 1:1 [9]. The heart secretes PICP into the peripheral circulation through the coronary sinus [9]. However, whether there is a positive correlation between plasma PICP, and myocardial collagen content remains controversial. A cross-sectional study demonstrated that plasma PICP levels in HCM patients were positively correlated with myocardial PICP content and the histological myocardial collagen volume fraction [10]. Similarly, Ferreira et al. also confirmed that the serum levels of PICP were significantly higher in patients with hypertension before drug treatment [11]. In contrast, in heart failure rats, Adamcova et al. observed that collagen content in the left ventricle was increased, whereas plasma PICP levels were reduced by 42% as compared to the control group [12]. These findings suggest that the myocardial collagen content does not necessarily correlate with plasma PICP, which may be related to confounding factors (such as weight loss and catabolic state). For predictive value, Ruiz-Ruiz et al. considered that serum PICP can be used independently for predicting HF episodes, hospital readmission, and death [13]. They included 111 patients with decompensated HF and found that 22.52% of the patients died during the 21 months of follow-up, and 48.6% of the patients were readmitted for HF. Moreover, serum PICP levels were significantly increased among patients who reached some primary endpoints (new hospitalizations or death) during follow-up (88.12 ± 37.31 ng/mL vs 73.13 ± 34.06 ng/mL; p = 0.029), and a cut-off value of 124 ng/mL predicted prognosis most accurately. Unfortunately, they did not perform cardiac biopsies, which is considered the most reliable method for measuring myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Propeptide of procollagen type I (PIP) and outcomes in decompensated heart failure
2
2007
... Type I collagen is a heterotrimeric molecule, which is composed of two α 1 chains and one α 2 chain. During its synthesis, the protein undergoes a series of post-translational modifications to form the procollagen chain; this precursor is then secreted into the extracellular space and cleaved by specific proteinases [8]. The stoichiometric ratio of the C-terminal propeptide of procollagen type I (PICP), which is released into the blood, and collagen type I produced by cleavage is 1:1 [9]. The heart secretes PICP into the peripheral circulation through the coronary sinus [9]. However, whether there is a positive correlation between plasma PICP, and myocardial collagen content remains controversial. A cross-sectional study demonstrated that plasma PICP levels in HCM patients were positively correlated with myocardial PICP content and the histological myocardial collagen volume fraction [10]. Similarly, Ferreira et al. also confirmed that the serum levels of PICP were significantly higher in patients with hypertension before drug treatment [11]. In contrast, in heart failure rats, Adamcova et al. observed that collagen content in the left ventricle was increased, whereas plasma PICP levels were reduced by 42% as compared to the control group [12]. These findings suggest that the myocardial collagen content does not necessarily correlate with plasma PICP, which may be related to confounding factors (such as weight loss and catabolic state). For predictive value, Ruiz-Ruiz et al. considered that serum PICP can be used independently for predicting HF episodes, hospital readmission, and death [13]. They included 111 patients with decompensated HF and found that 22.52% of the patients died during the 21 months of follow-up, and 48.6% of the patients were readmitted for HF. Moreover, serum PICP levels were significantly increased among patients who reached some primary endpoints (new hospitalizations or death) during follow-up (88.12 ± 37.31 ng/mL vs 73.13 ± 34.06 ng/mL; p = 0.029), and a cut-off value of 124 ng/mL predicted prognosis most accurately. Unfortunately, they did not perform cardiac biopsies, which is considered the most reliable method for measuring myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Myocardial fibrosis in hypertensive heart disease: an overview of potential regulatory mechanisms
1
1995
... Excessive deposition of collagen type I is a feature of cardiac remodeling [14]. The procollagen type I N-terminal propeptide (PINP) originates from conversion of collagen type I, and is a marker for collagen type I synthesis [15]. N-terminal pro-peptides are removed by members of the ADAMTS family [16]. Compared with PICP, PINP has the disadvantage of delayed release [17]. At present, the relationship between PINP levels and fibrosis remains elusive. In a case?control study, Zile et al. observed that the baseline serum PINP levels in patients with HF were significantly higher than those in controls [18]. In rats with ischemic cardiomyopathy, Zhang et al. detected significant cardiac fibrosis by Masson staining 6 weeks after myocardial infarction (MI), and enzyme-linked immunosorbent assay (ELISA) results showed that the level of plasma PINP in the MI group was significantly increased as compared to that in the normal control group [19]. However, in a cross-sectional study in which serum PINP levels were measured by radioimmunoassay, no significant difference was revealed between HCM patients and healthy individuals [20]. As shown by the above studies, PINP has some limitations as a biomarker. ...
Radioimmunoassay of the carboxyterminal propeptide of human type I procollagen
1
1990
... Excessive deposition of collagen type I is a feature of cardiac remodeling [14]. The procollagen type I N-terminal propeptide (PINP) originates from conversion of collagen type I, and is a marker for collagen type I synthesis [15]. N-terminal pro-peptides are removed by members of the ADAMTS family [16]. Compared with PICP, PINP has the disadvantage of delayed release [17]. At present, the relationship between PINP levels and fibrosis remains elusive. In a case?control study, Zile et al. observed that the baseline serum PINP levels in patients with HF were significantly higher than those in controls [18]. In rats with ischemic cardiomyopathy, Zhang et al. detected significant cardiac fibrosis by Masson staining 6 weeks after myocardial infarction (MI), and enzyme-linked immunosorbent assay (ELISA) results showed that the level of plasma PINP in the MI group was significantly increased as compared to that in the normal control group [19]. However, in a cross-sectional study in which serum PINP levels were measured by radioimmunoassay, no significant difference was revealed between HCM patients and healthy individuals [20]. As shown by the above studies, PINP has some limitations as a biomarker. ...
Cloning and characterization of ADAMTS-14, a novel ADAMTS displaying high homology with ADAMTS-2 and ADAMTS-3
1
2002
... Excessive deposition of collagen type I is a feature of cardiac remodeling [14]. The procollagen type I N-terminal propeptide (PINP) originates from conversion of collagen type I, and is a marker for collagen type I synthesis [15]. N-terminal pro-peptides are removed by members of the ADAMTS family [16]. Compared with PICP, PINP has the disadvantage of delayed release [17]. At present, the relationship between PINP levels and fibrosis remains elusive. In a case?control study, Zile et al. observed that the baseline serum PINP levels in patients with HF were significantly higher than those in controls [18]. In rats with ischemic cardiomyopathy, Zhang et al. detected significant cardiac fibrosis by Masson staining 6 weeks after myocardial infarction (MI), and enzyme-linked immunosorbent assay (ELISA) results showed that the level of plasma PINP in the MI group was significantly increased as compared to that in the normal control group [19]. However, in a cross-sectional study in which serum PINP levels were measured by radioimmunoassay, no significant difference was revealed between HCM patients and healthy individuals [20]. As shown by the above studies, PINP has some limitations as a biomarker. ...
Analysing connective tissue metabolites in human serum. Biochemical, physiological and methodological aspects
2
1995
... Excessive deposition of collagen type I is a feature of cardiac remodeling [14]. The procollagen type I N-terminal propeptide (PINP) originates from conversion of collagen type I, and is a marker for collagen type I synthesis [15]. N-terminal pro-peptides are removed by members of the ADAMTS family [16]. Compared with PICP, PINP has the disadvantage of delayed release [17]. At present, the relationship between PINP levels and fibrosis remains elusive. In a case?control study, Zile et al. observed that the baseline serum PINP levels in patients with HF were significantly higher than those in controls [18]. In rats with ischemic cardiomyopathy, Zhang et al. detected significant cardiac fibrosis by Masson staining 6 weeks after myocardial infarction (MI), and enzyme-linked immunosorbent assay (ELISA) results showed that the level of plasma PINP in the MI group was significantly increased as compared to that in the normal control group [19]. However, in a cross-sectional study in which serum PINP levels were measured by radioimmunoassay, no significant difference was revealed between HCM patients and healthy individuals [20]. As shown by the above studies, PINP has some limitations as a biomarker. ...
... Collagen type III, whose fibers have a relatively small diameter, is synthesized by cardiac fibroblasts and is mainly responsible for myocardial elasticity [21, 22]. After the C-propeptides of fibrillar procollagen are cleaved by proteases, procollagen type III amino-terminal propeptide (PIIINP) is released into the blood via the lymphatics [17]. The N-propeptide is considered to play an important role in the regulation of primary fibril diameter [23]. Excellent storage and stability are the characteristics of PIIINP [24]. However, PIIINP is sometimes not completely separated from procollagen, which leads to the underestimation of type III collagen synthesis. In a prospective study of dilated cardiomyopathy, baseline serum PIIINP levels in patients with idiopathic or ischemic dilated cardiomyopathy were significantly higher than those in healthy controls, and serum PIIINP levels were highly correlated with cardiac collagen type III levels [25]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis. ...
Effects of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients With HFrEF
2
2019
... Excessive deposition of collagen type I is a feature of cardiac remodeling [14]. The procollagen type I N-terminal propeptide (PINP) originates from conversion of collagen type I, and is a marker for collagen type I synthesis [15]. N-terminal pro-peptides are removed by members of the ADAMTS family [16]. Compared with PICP, PINP has the disadvantage of delayed release [17]. At present, the relationship between PINP levels and fibrosis remains elusive. In a case?control study, Zile et al. observed that the baseline serum PINP levels in patients with HF were significantly higher than those in controls [18]. In rats with ischemic cardiomyopathy, Zhang et al. detected significant cardiac fibrosis by Masson staining 6 weeks after myocardial infarction (MI), and enzyme-linked immunosorbent assay (ELISA) results showed that the level of plasma PINP in the MI group was significantly increased as compared to that in the normal control group [19]. However, in a cross-sectional study in which serum PINP levels were measured by radioimmunoassay, no significant difference was revealed between HCM patients and healthy individuals [20]. As shown by the above studies, PINP has some limitations as a biomarker. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Renal Denervation Effects on Myocardial Fibrosis and Ventricular Arrhythmias in Rats with Ischemic Cardiomyopathy
2
2018
... Excessive deposition of collagen type I is a feature of cardiac remodeling [14]. The procollagen type I N-terminal propeptide (PINP) originates from conversion of collagen type I, and is a marker for collagen type I synthesis [15]. N-terminal pro-peptides are removed by members of the ADAMTS family [16]. Compared with PICP, PINP has the disadvantage of delayed release [17]. At present, the relationship between PINP levels and fibrosis remains elusive. In a case?control study, Zile et al. observed that the baseline serum PINP levels in patients with HF were significantly higher than those in controls [18]. In rats with ischemic cardiomyopathy, Zhang et al. detected significant cardiac fibrosis by Masson staining 6 weeks after myocardial infarction (MI), and enzyme-linked immunosorbent assay (ELISA) results showed that the level of plasma PINP in the MI group was significantly increased as compared to that in the normal control group [19]. However, in a cross-sectional study in which serum PINP levels were measured by radioimmunoassay, no significant difference was revealed between HCM patients and healthy individuals [20]. As shown by the above studies, PINP has some limitations as a biomarker. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Myocardial collagen turnover in hypertrophic cardiomyopathy
4
2003
... Excessive deposition of collagen type I is a feature of cardiac remodeling [14]. The procollagen type I N-terminal propeptide (PINP) originates from conversion of collagen type I, and is a marker for collagen type I synthesis [15]. N-terminal pro-peptides are removed by members of the ADAMTS family [16]. Compared with PICP, PINP has the disadvantage of delayed release [17]. At present, the relationship between PINP levels and fibrosis remains elusive. In a case?control study, Zile et al. observed that the baseline serum PINP levels in patients with HF were significantly higher than those in controls [18]. In rats with ischemic cardiomyopathy, Zhang et al. detected significant cardiac fibrosis by Masson staining 6 weeks after myocardial infarction (MI), and enzyme-linked immunosorbent assay (ELISA) results showed that the level of plasma PINP in the MI group was significantly increased as compared to that in the normal control group [19]. However, in a cross-sectional study in which serum PINP levels were measured by radioimmunoassay, no significant difference was revealed between HCM patients and healthy individuals [20]. As shown by the above studies, PINP has some limitations as a biomarker. ...
... During degradation of collagen type I fibrils, the C-terminal telopeptide of collagen type I (CITP) is a cross-linked terminal peptide released in a 1:1 stoichiometric ratio, allowing accurate measurement of collagen degradation [30, 31]. In a cross-sectional study, serum CITP levels in HCM patients were increased, while the PICP and PINP levels were not altered significantly, indicating a shift in the collagen balance toward collagen type I breakdown [20]. This is an important outcome, because increased myocardial stiffness is usually caused by collagen deposition. However, several other studies have shown that the relationship between CITP and myocardial fibrosis is controversial. In patients with HF and atrial fibrillation (AF), serum CITP levels were significantly higher than those in the control groups [32, 33]. But Nagao et al. observed that serum CITP levels in patients with DCM were not associated with left ventricular remodeling parameters, or with the expression of cardiac collagen type I and type III [34]. In addition, some studies have confirmed the predictive value of CITP. Manhenke et al. considered that plasma CITP was an independent predictor of cardiovascular mortality in patients with acute myocardial infarction (AMI) [35]. This prospective study included 233 patients with AMI, among whom 56% reached the combined endpoint of HF symptoms or CV death during the years of follow-up, and plasma ICTP was increased in patients who died due to any cause. Similarly, serum CITP is also useful for determining cardiac events in patients with complete HF [36]. In summary, CITP may facilitate diagnosis or prognosis of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Semiquantitative analysis of collagen types in the hypertrophied left ventricle
1
2001
... Collagen type III, whose fibers have a relatively small diameter, is synthesized by cardiac fibroblasts and is mainly responsible for myocardial elasticity [21, 22]. After the C-propeptides of fibrillar procollagen are cleaved by proteases, procollagen type III amino-terminal propeptide (PIIINP) is released into the blood via the lymphatics [17]. The N-propeptide is considered to play an important role in the regulation of primary fibril diameter [23]. Excellent storage and stability are the characteristics of PIIINP [24]. However, PIIINP is sometimes not completely separated from procollagen, which leads to the underestimation of type III collagen synthesis. In a prospective study of dilated cardiomyopathy, baseline serum PIIINP levels in patients with idiopathic or ischemic dilated cardiomyopathy were significantly higher than those in healthy controls, and serum PIIINP levels were highly correlated with cardiac collagen type III levels [25]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis. ...
The relationship between myocardial extracellular matrix remodeling and ventricular function
1
2006
... Collagen type III, whose fibers have a relatively small diameter, is synthesized by cardiac fibroblasts and is mainly responsible for myocardial elasticity [21, 22]. After the C-propeptides of fibrillar procollagen are cleaved by proteases, procollagen type III amino-terminal propeptide (PIIINP) is released into the blood via the lymphatics [17]. The N-propeptide is considered to play an important role in the regulation of primary fibril diameter [23]. Excellent storage and stability are the characteristics of PIIINP [24]. However, PIIINP is sometimes not completely separated from procollagen, which leads to the underestimation of type III collagen synthesis. In a prospective study of dilated cardiomyopathy, baseline serum PIIINP levels in patients with idiopathic or ischemic dilated cardiomyopathy were significantly higher than those in healthy controls, and serum PIIINP levels were highly correlated with cardiac collagen type III levels [25]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis. ...
Collagens--structure, function, and biosynthesis
1
2003
... Collagen type III, whose fibers have a relatively small diameter, is synthesized by cardiac fibroblasts and is mainly responsible for myocardial elasticity [21, 22]. After the C-propeptides of fibrillar procollagen are cleaved by proteases, procollagen type III amino-terminal propeptide (PIIINP) is released into the blood via the lymphatics [17]. The N-propeptide is considered to play an important role in the regulation of primary fibril diameter [23]. Excellent storage and stability are the characteristics of PIIINP [24]. However, PIIINP is sometimes not completely separated from procollagen, which leads to the underestimation of type III collagen synthesis. In a prospective study of dilated cardiomyopathy, baseline serum PIIINP levels in patients with idiopathic or ischemic dilated cardiomyopathy were significantly higher than those in healthy controls, and serum PIIINP levels were highly correlated with cardiac collagen type III levels [25]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis. ...
The effects of a freeze-thaw cycle and pre-analytical storage temperature on the stability of insulin-like growth factor-I and pro-collagen type III N-terminal propeptide concentrations: Implications for the detection of growth hormone misuse in athletes
1
2012
... Collagen type III, whose fibers have a relatively small diameter, is synthesized by cardiac fibroblasts and is mainly responsible for myocardial elasticity [21, 22]. After the C-propeptides of fibrillar procollagen are cleaved by proteases, procollagen type III amino-terminal propeptide (PIIINP) is released into the blood via the lymphatics [17]. The N-propeptide is considered to play an important role in the regulation of primary fibril diameter [23]. Excellent storage and stability are the characteristics of PIIINP [24]. However, PIIINP is sometimes not completely separated from procollagen, which leads to the underestimation of type III collagen synthesis. In a prospective study of dilated cardiomyopathy, baseline serum PIIINP levels in patients with idiopathic or ischemic dilated cardiomyopathy were significantly higher than those in healthy controls, and serum PIIINP levels were highly correlated with cardiac collagen type III levels [25]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis. ...
Measuring extracellular matrix turnover in the serum of patients with idiopathic or ischemic dilated cardiomyopathy and impact on diagnosis and prognosis
4
1995
... Collagen type III, whose fibers have a relatively small diameter, is synthesized by cardiac fibroblasts and is mainly responsible for myocardial elasticity [21, 22]. After the C-propeptides of fibrillar procollagen are cleaved by proteases, procollagen type III amino-terminal propeptide (PIIINP) is released into the blood via the lymphatics [17]. The N-propeptide is considered to play an important role in the regulation of primary fibril diameter [23]. Excellent storage and stability are the characteristics of PIIINP [24]. However, PIIINP is sometimes not completely separated from procollagen, which leads to the underestimation of type III collagen synthesis. In a prospective study of dilated cardiomyopathy, baseline serum PIIINP levels in patients with idiopathic or ischemic dilated cardiomyopathy were significantly higher than those in healthy controls, and serum PIIINP levels were highly correlated with cardiac collagen type III levels [25]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis. ...
... ]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
... It is important to note that fibrosis occurs not only in the heart, but also in other organs, so that changes in biomarker levels may not have only a cardiac origin [25]. In another respect, biomarkers must be strictly tested to determine whether they strongly reflect myocardial fibrosis. Endomyocardial biopsy can be used to estimate the usefulness and accuracy of biomarkers. Only when these initiatives are successful can biomarkers be incorporated into clinical practice. Additionally, cost-effectivity issues should also be taken into consideration, as the measurement of many biomarkers mentioned is not cheap, particularly when using a multiple-biomarker approach. Moreover, there is not currently a well-tested biomarker for fibrosis that is equivalent to NT-proBNP for HF. On the whole, the use of biomarkers is helpful in the assessment of myocardial fibrosis; therefore, more prospective studies are needed in the future. ...
The relationship between serum fibrosis markers and restrictive ventricular filling in patients with heart failure with reduced ejection fraction: A technetium-99m radionuclide ventriculography study
2
2017
... Collagen type III, whose fibers have a relatively small diameter, is synthesized by cardiac fibroblasts and is mainly responsible for myocardial elasticity [21, 22]. After the C-propeptides of fibrillar procollagen are cleaved by proteases, procollagen type III amino-terminal propeptide (PIIINP) is released into the blood via the lymphatics [17]. The N-propeptide is considered to play an important role in the regulation of primary fibril diameter [23]. Excellent storage and stability are the characteristics of PIIINP [24]. However, PIIINP is sometimes not completely separated from procollagen, which leads to the underestimation of type III collagen synthesis. In a prospective study of dilated cardiomyopathy, baseline serum PIIINP levels in patients with idiopathic or ischemic dilated cardiomyopathy were significantly higher than those in healthy controls, and serum PIIINP levels were highly correlated with cardiac collagen type III levels [25]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Serum concentration of procollagen type III amino-terminal peptide is increased in patients with successfully repaired coarctation of the aorta with left ventricular hypertrophy
2
2015
... Collagen type III, whose fibers have a relatively small diameter, is synthesized by cardiac fibroblasts and is mainly responsible for myocardial elasticity [21, 22]. After the C-propeptides of fibrillar procollagen are cleaved by proteases, procollagen type III amino-terminal propeptide (PIIINP) is released into the blood via the lymphatics [17]. The N-propeptide is considered to play an important role in the regulation of primary fibril diameter [23]. Excellent storage and stability are the characteristics of PIIINP [24]. However, PIIINP is sometimes not completely separated from procollagen, which leads to the underestimation of type III collagen synthesis. In a prospective study of dilated cardiomyopathy, baseline serum PIIINP levels in patients with idiopathic or ischemic dilated cardiomyopathy were significantly higher than those in healthy controls, and serum PIIINP levels were highly correlated with cardiac collagen type III levels [25]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Procollagen type III amino terminal peptide and myocardial fibrosis: A study in hypertensive patients with and without left ventricular hypertrophy
2
2015
... Collagen type III, whose fibers have a relatively small diameter, is synthesized by cardiac fibroblasts and is mainly responsible for myocardial elasticity [21, 22]. After the C-propeptides of fibrillar procollagen are cleaved by proteases, procollagen type III amino-terminal propeptide (PIIINP) is released into the blood via the lymphatics [17]. The N-propeptide is considered to play an important role in the regulation of primary fibril diameter [23]. Excellent storage and stability are the characteristics of PIIINP [24]. However, PIIINP is sometimes not completely separated from procollagen, which leads to the underestimation of type III collagen synthesis. In a prospective study of dilated cardiomyopathy, baseline serum PIIINP levels in patients with idiopathic or ischemic dilated cardiomyopathy were significantly higher than those in healthy controls, and serum PIIINP levels were highly correlated with cardiac collagen type III levels [25]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Tanshinone IIA prevents left ventricular remodelling via the TLR4/MyD88/NF-κB signalling pathway in rats with myocardial infarction
2
2018
... Collagen type III, whose fibers have a relatively small diameter, is synthesized by cardiac fibroblasts and is mainly responsible for myocardial elasticity [21, 22]. After the C-propeptides of fibrillar procollagen are cleaved by proteases, procollagen type III amino-terminal propeptide (PIIINP) is released into the blood via the lymphatics [17]. The N-propeptide is considered to play an important role in the regulation of primary fibril diameter [23]. Excellent storage and stability are the characteristics of PIIINP [24]. However, PIIINP is sometimes not completely separated from procollagen, which leads to the underestimation of type III collagen synthesis. In a prospective study of dilated cardiomyopathy, baseline serum PIIINP levels in patients with idiopathic or ischemic dilated cardiomyopathy were significantly higher than those in healthy controls, and serum PIIINP levels were highly correlated with cardiac collagen type III levels [25]. Patients with a serum PIIINP value > 7 pg/L had a higher risk of a poor hemodynamic condition, advanced clinical stage, heart transplantation, hyponatremia, and death during follow-up than did patients with low PIIINP values [25]. This shows that the increase in serum PIIINP levels could reflect cardiac fibrosis to some extent, which was of prognostic value and was related to clinical stage. Additionally, PIIINP levels were positively correlated with diastolic dysfunction in patients with HF with reduced ejection fraction (HFrEF) [26], with left ventricular mass index (LVMI), and with relative wall thickness (RWT) in patients with successfully repaired coarctation of the aorta (CoA) with left ventricular hypertrophy [27]. It was also inversely associated with diastolic function in patients with hypertension [28]. Moreover, in myocardial infarction (MI) model rats, the reduction in fibrosis with tanshinone IIA was accompanied by a reduction in serum PIIINP [29]. Taken together, PIIINP can reflect the degree of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Dynamic state of collagen: pathways of collagen degradation in vivo and their possible role in regulation of collagen mass
1
1987
... During degradation of collagen type I fibrils, the C-terminal telopeptide of collagen type I (CITP) is a cross-linked terminal peptide released in a 1:1 stoichiometric ratio, allowing accurate measurement of collagen degradation [30, 31]. In a cross-sectional study, serum CITP levels in HCM patients were increased, while the PICP and PINP levels were not altered significantly, indicating a shift in the collagen balance toward collagen type I breakdown [20]. This is an important outcome, because increased myocardial stiffness is usually caused by collagen deposition. However, several other studies have shown that the relationship between CITP and myocardial fibrosis is controversial. In patients with HF and atrial fibrillation (AF), serum CITP levels were significantly higher than those in the control groups [32, 33]. But Nagao et al. observed that serum CITP levels in patients with DCM were not associated with left ventricular remodeling parameters, or with the expression of cardiac collagen type I and type III [34]. In addition, some studies have confirmed the predictive value of CITP. Manhenke et al. considered that plasma CITP was an independent predictor of cardiovascular mortality in patients with acute myocardial infarction (AMI) [35]. This prospective study included 233 patients with AMI, among whom 56% reached the combined endpoint of HF symptoms or CV death during the years of follow-up, and plasma ICTP was increased in patients who died due to any cause. Similarly, serum CITP is also useful for determining cardiac events in patients with complete HF [36]. In summary, CITP may facilitate diagnosis or prognosis of myocardial fibrosis. ...
Radioimmunoassay for the pyridinoline cross-linked carboxy-terminal telopeptide of type I collagen: A new serum marker of bone collagen degradation
1
1993
... During degradation of collagen type I fibrils, the C-terminal telopeptide of collagen type I (CITP) is a cross-linked terminal peptide released in a 1:1 stoichiometric ratio, allowing accurate measurement of collagen degradation [30, 31]. In a cross-sectional study, serum CITP levels in HCM patients were increased, while the PICP and PINP levels were not altered significantly, indicating a shift in the collagen balance toward collagen type I breakdown [20]. This is an important outcome, because increased myocardial stiffness is usually caused by collagen deposition. However, several other studies have shown that the relationship between CITP and myocardial fibrosis is controversial. In patients with HF and atrial fibrillation (AF), serum CITP levels were significantly higher than those in the control groups [32, 33]. But Nagao et al. observed that serum CITP levels in patients with DCM were not associated with left ventricular remodeling parameters, or with the expression of cardiac collagen type I and type III [34]. In addition, some studies have confirmed the predictive value of CITP. Manhenke et al. considered that plasma CITP was an independent predictor of cardiovascular mortality in patients with acute myocardial infarction (AMI) [35]. This prospective study included 233 patients with AMI, among whom 56% reached the combined endpoint of HF symptoms or CV death during the years of follow-up, and plasma ICTP was increased in patients who died due to any cause. Similarly, serum CITP is also useful for determining cardiac events in patients with complete HF [36]. In summary, CITP may facilitate diagnosis or prognosis of myocardial fibrosis. ...
Circulating multimarker profile of patients with symptomatic heart failure supports enhanced fibrotic degradation and decreased angiogenesis
2
2016
... During degradation of collagen type I fibrils, the C-terminal telopeptide of collagen type I (CITP) is a cross-linked terminal peptide released in a 1:1 stoichiometric ratio, allowing accurate measurement of collagen degradation [30, 31]. In a cross-sectional study, serum CITP levels in HCM patients were increased, while the PICP and PINP levels were not altered significantly, indicating a shift in the collagen balance toward collagen type I breakdown [20]. This is an important outcome, because increased myocardial stiffness is usually caused by collagen deposition. However, several other studies have shown that the relationship between CITP and myocardial fibrosis is controversial. In patients with HF and atrial fibrillation (AF), serum CITP levels were significantly higher than those in the control groups [32, 33]. But Nagao et al. observed that serum CITP levels in patients with DCM were not associated with left ventricular remodeling parameters, or with the expression of cardiac collagen type I and type III [34]. In addition, some studies have confirmed the predictive value of CITP. Manhenke et al. considered that plasma CITP was an independent predictor of cardiovascular mortality in patients with acute myocardial infarction (AMI) [35]. This prospective study included 233 patients with AMI, among whom 56% reached the combined endpoint of HF symptoms or CV death during the years of follow-up, and plasma ICTP was increased in patients who died due to any cause. Similarly, serum CITP is also useful for determining cardiac events in patients with complete HF [36]. In summary, CITP may facilitate diagnosis or prognosis of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Extracellular matrix alterations in patients with paroxysmal and persistent atrial fibrillation: biochemical assessment of collagen type-I turnover
2
2008
... During degradation of collagen type I fibrils, the C-terminal telopeptide of collagen type I (CITP) is a cross-linked terminal peptide released in a 1:1 stoichiometric ratio, allowing accurate measurement of collagen degradation [30, 31]. In a cross-sectional study, serum CITP levels in HCM patients were increased, while the PICP and PINP levels were not altered significantly, indicating a shift in the collagen balance toward collagen type I breakdown [20]. This is an important outcome, because increased myocardial stiffness is usually caused by collagen deposition. However, several other studies have shown that the relationship between CITP and myocardial fibrosis is controversial. In patients with HF and atrial fibrillation (AF), serum CITP levels were significantly higher than those in the control groups [32, 33]. But Nagao et al. observed that serum CITP levels in patients with DCM were not associated with left ventricular remodeling parameters, or with the expression of cardiac collagen type I and type III [34]. In addition, some studies have confirmed the predictive value of CITP. Manhenke et al. considered that plasma CITP was an independent predictor of cardiovascular mortality in patients with acute myocardial infarction (AMI) [35]. This prospective study included 233 patients with AMI, among whom 56% reached the combined endpoint of HF symptoms or CV death during the years of follow-up, and plasma ICTP was increased in patients who died due to any cause. Similarly, serum CITP is also useful for determining cardiac events in patients with complete HF [36]. In summary, CITP may facilitate diagnosis or prognosis of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Circulating markers of collagen types I, III, and IV in patients with dilated cardiomyopathy: relationships with myocardial collagen expression
2
2018
... During degradation of collagen type I fibrils, the C-terminal telopeptide of collagen type I (CITP) is a cross-linked terminal peptide released in a 1:1 stoichiometric ratio, allowing accurate measurement of collagen degradation [30, 31]. In a cross-sectional study, serum CITP levels in HCM patients were increased, while the PICP and PINP levels were not altered significantly, indicating a shift in the collagen balance toward collagen type I breakdown [20]. This is an important outcome, because increased myocardial stiffness is usually caused by collagen deposition. However, several other studies have shown that the relationship between CITP and myocardial fibrosis is controversial. In patients with HF and atrial fibrillation (AF), serum CITP levels were significantly higher than those in the control groups [32, 33]. But Nagao et al. observed that serum CITP levels in patients with DCM were not associated with left ventricular remodeling parameters, or with the expression of cardiac collagen type I and type III [34]. In addition, some studies have confirmed the predictive value of CITP. Manhenke et al. considered that plasma CITP was an independent predictor of cardiovascular mortality in patients with acute myocardial infarction (AMI) [35]. This prospective study included 233 patients with AMI, among whom 56% reached the combined endpoint of HF symptoms or CV death during the years of follow-up, and plasma ICTP was increased in patients who died due to any cause. Similarly, serum CITP is also useful for determining cardiac events in patients with complete HF [36]. In summary, CITP may facilitate diagnosis or prognosis of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
The prognostic value of circulating markers of collagen turnover after acute myocardial infarction
2
2011
... During degradation of collagen type I fibrils, the C-terminal telopeptide of collagen type I (CITP) is a cross-linked terminal peptide released in a 1:1 stoichiometric ratio, allowing accurate measurement of collagen degradation [30, 31]. In a cross-sectional study, serum CITP levels in HCM patients were increased, while the PICP and PINP levels were not altered significantly, indicating a shift in the collagen balance toward collagen type I breakdown [20]. This is an important outcome, because increased myocardial stiffness is usually caused by collagen deposition. However, several other studies have shown that the relationship between CITP and myocardial fibrosis is controversial. In patients with HF and atrial fibrillation (AF), serum CITP levels were significantly higher than those in the control groups [32, 33]. But Nagao et al. observed that serum CITP levels in patients with DCM were not associated with left ventricular remodeling parameters, or with the expression of cardiac collagen type I and type III [34]. In addition, some studies have confirmed the predictive value of CITP. Manhenke et al. considered that plasma CITP was an independent predictor of cardiovascular mortality in patients with acute myocardial infarction (AMI) [35]. This prospective study included 233 patients with AMI, among whom 56% reached the combined endpoint of HF symptoms or CV death during the years of follow-up, and plasma ICTP was increased in patients who died due to any cause. Similarly, serum CITP is also useful for determining cardiac events in patients with complete HF [36]. In summary, CITP may facilitate diagnosis or prognosis of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Serum carboxy-terminal telopeptide of type I collagen (ICTP) predicts cardiac events in chronic heart failure patients with preserved left ventricular systolic function
1
2007
... During degradation of collagen type I fibrils, the C-terminal telopeptide of collagen type I (CITP) is a cross-linked terminal peptide released in a 1:1 stoichiometric ratio, allowing accurate measurement of collagen degradation [30, 31]. In a cross-sectional study, serum CITP levels in HCM patients were increased, while the PICP and PINP levels were not altered significantly, indicating a shift in the collagen balance toward collagen type I breakdown [20]. This is an important outcome, because increased myocardial stiffness is usually caused by collagen deposition. However, several other studies have shown that the relationship between CITP and myocardial fibrosis is controversial. In patients with HF and atrial fibrillation (AF), serum CITP levels were significantly higher than those in the control groups [32, 33]. But Nagao et al. observed that serum CITP levels in patients with DCM were not associated with left ventricular remodeling parameters, or with the expression of cardiac collagen type I and type III [34]. In addition, some studies have confirmed the predictive value of CITP. Manhenke et al. considered that plasma CITP was an independent predictor of cardiovascular mortality in patients with acute myocardial infarction (AMI) [35]. This prospective study included 233 patients with AMI, among whom 56% reached the combined endpoint of HF symptoms or CV death during the years of follow-up, and plasma ICTP was increased in patients who died due to any cause. Similarly, serum CITP is also useful for determining cardiac events in patients with complete HF [36]. In summary, CITP may facilitate diagnosis or prognosis of myocardial fibrosis. ...
Structure and function of matrix metalloproteinases and TIMPs
1
2006
... Matrix metalloproteinases (MMPs) are zinc- and calcium-dependent peptide enzymes that are involved in myocardial remodeling and degradation of collagen [37, 38]. It promotes the degradation of extracellular matrix, such as collagen, elastin, and gelatin [39]. MMPs can hydrolyze collagen efficiently, but their relative activities towards interstitial collagens are different. At 25°C, MMP-1 preferentially cleaves collagen type III over types I and II [40]. MMP-13 cleaves collagen type II five-fold faster than collagen type I and six-fold more rapidly than collagen type III, at 25 °C [41]. For MMP-2 and MMP-9, collagen type III is the substrate preferred over collagen types I and II [42, 43]. ...
TIMP-1: a marker of left ventricular diastolic dysfunction and fibrosis in hypertension
1
2002
... Matrix metalloproteinases (MMPs) are zinc- and calcium-dependent peptide enzymes that are involved in myocardial remodeling and degradation of collagen [37, 38]. It promotes the degradation of extracellular matrix, such as collagen, elastin, and gelatin [39]. MMPs can hydrolyze collagen efficiently, but their relative activities towards interstitial collagens are different. At 25°C, MMP-1 preferentially cleaves collagen type III over types I and II [40]. MMP-13 cleaves collagen type II five-fold faster than collagen type I and six-fold more rapidly than collagen type III, at 25 °C [41]. For MMP-2 and MMP-9, collagen type III is the substrate preferred over collagen types I and II [42, 43]. ...
Biochemical and Biological Attributes of Matrix Metalloproteinases
1
2017
... Matrix metalloproteinases (MMPs) are zinc- and calcium-dependent peptide enzymes that are involved in myocardial remodeling and degradation of collagen [37, 38]. It promotes the degradation of extracellular matrix, such as collagen, elastin, and gelatin [39]. MMPs can hydrolyze collagen efficiently, but their relative activities towards interstitial collagens are different. At 25°C, MMP-1 preferentially cleaves collagen type III over types I and II [40]. MMP-13 cleaves collagen type II five-fold faster than collagen type I and six-fold more rapidly than collagen type III, at 25 °C [41]. For MMP-2 and MMP-9, collagen type III is the substrate preferred over collagen types I and II [42, 43]. ...
The collagen substrate specificity of human neutrophil collagenase
1
1987
... Matrix metalloproteinases (MMPs) are zinc- and calcium-dependent peptide enzymes that are involved in myocardial remodeling and degradation of collagen [37, 38]. It promotes the degradation of extracellular matrix, such as collagen, elastin, and gelatin [39]. MMPs can hydrolyze collagen efficiently, but their relative activities towards interstitial collagens are different. At 25°C, MMP-1 preferentially cleaves collagen type III over types I and II [40]. MMP-13 cleaves collagen type II five-fold faster than collagen type I and six-fold more rapidly than collagen type III, at 25 °C [41]. For MMP-2 and MMP-9, collagen type III is the substrate preferred over collagen types I and II [42, 43]. ...
Biochemical characterization of human collagenase-3
1
1996
... Matrix metalloproteinases (MMPs) are zinc- and calcium-dependent peptide enzymes that are involved in myocardial remodeling and degradation of collagen [37, 38]. It promotes the degradation of extracellular matrix, such as collagen, elastin, and gelatin [39]. MMPs can hydrolyze collagen efficiently, but their relative activities towards interstitial collagens are different. At 25°C, MMP-1 preferentially cleaves collagen type III over types I and II [40]. MMP-13 cleaves collagen type II five-fold faster than collagen type I and six-fold more rapidly than collagen type III, at 25 °C [41]. For MMP-2 and MMP-9, collagen type III is the substrate preferred over collagen types I and II [42, 43]. ...
New collagenolytic enzymes/cascade identified at the pannus-hard tissue junction in rheumatoid arthritis: destruction from above
1
1998
... Matrix metalloproteinases (MMPs) are zinc- and calcium-dependent peptide enzymes that are involved in myocardial remodeling and degradation of collagen [37, 38]. It promotes the degradation of extracellular matrix, such as collagen, elastin, and gelatin [39]. MMPs can hydrolyze collagen efficiently, but their relative activities towards interstitial collagens are different. At 25°C, MMP-1 preferentially cleaves collagen type III over types I and II [40]. MMP-13 cleaves collagen type II five-fold faster than collagen type I and six-fold more rapidly than collagen type III, at 25 °C [41]. For MMP-2 and MMP-9, collagen type III is the substrate preferred over collagen types I and II [42, 43]. ...
Activity of matrix metalloproteinase-9 against native collagen types I and III
1
2007
... Matrix metalloproteinases (MMPs) are zinc- and calcium-dependent peptide enzymes that are involved in myocardial remodeling and degradation of collagen [37, 38]. It promotes the degradation of extracellular matrix, such as collagen, elastin, and gelatin [39]. MMPs can hydrolyze collagen efficiently, but their relative activities towards interstitial collagens are different. At 25°C, MMP-1 preferentially cleaves collagen type III over types I and II [40]. MMP-13 cleaves collagen type II five-fold faster than collagen type I and six-fold more rapidly than collagen type III, at 25 °C [41]. For MMP-2 and MMP-9, collagen type III is the substrate preferred over collagen types I and II [42, 43]. ...
Serum Matrix Metalloproteinases as Quantitative Biomarkers for Myocardial Fibrosis and Sudden Cardiac Death Risk Stratification in Patients With Hypertrophic Cardiomyopathy
3
2016
... Many MMPs are expressed in the myocardium. In HCM, the expression of MMPs is altered, but not all MMPs are affected. Münch et al. found that MMP-1, MMP-2, MMP-3, and MMP-9 are involved in HCM [44]. They observed that increased serum MMP-2 levels in females were associated with lower fibrosis, while MMP-9 was positively associated with fibrosis in late gadolinium enhancement cardiac magnetic resonance (mean increase of 0.66 g/unit MMP-9 [0.50;0.82], p < 0.001), but neither serum MMP-1 nor MMP-3 levels were associated with cardiac fibrosis [44]. However, a cautionary note on these results is that the cardiac tissue concentration of MMPs was not evaluated, which might present a limitation. Regarding MMP-1 levels in systolic and diastolic HF, serum MMP-1 levels were increased in systolic HF patients, but not in diastolic HF patients [45, 46]. This result reminds us that MMP activity also fluctuates because heart remodeling during HF is a dynamic process. In diabetic mice (DM), myocardial fibrosis is clearly related to the expression of MMP7, MMP11, MMP13, and MMP16 in myocardial tissue [47]. Compared with the control group, levels of MMP7, MMP11, MMP13, and MMP16 were markedly higher, but levels of MMP2 were markedly lower in the DM group [47]. It is also important to note the measurement method of MMPs in all the above studies. The levels of different MMPs are determined by immunohistochemistry or histology or by enzyme-linked immunosorbent assays, but these methods do not distinguish proMMPs from active MMPs. Therefore, these results are of limited value. ...
... ]. They observed that increased serum MMP-2 levels in females were associated with lower fibrosis, while MMP-9 was positively associated with fibrosis in late gadolinium enhancement cardiac magnetic resonance (mean increase of 0.66 g/unit MMP-9 [0.50;0.82], p < 0.001), but neither serum MMP-1 nor MMP-3 levels were associated with cardiac fibrosis [44]. However, a cautionary note on these results is that the cardiac tissue concentration of MMPs was not evaluated, which might present a limitation. Regarding MMP-1 levels in systolic and diastolic HF, serum MMP-1 levels were increased in systolic HF patients, but not in diastolic HF patients [45, 46]. This result reminds us that MMP activity also fluctuates because heart remodeling during HF is a dynamic process. In diabetic mice (DM), myocardial fibrosis is clearly related to the expression of MMP7, MMP11, MMP13, and MMP16 in myocardial tissue [47]. Compared with the control group, levels of MMP7, MMP11, MMP13, and MMP16 were markedly higher, but levels of MMP2 were markedly lower in the DM group [47]. It is also important to note the measurement method of MMPs in all the above studies. The levels of different MMPs are determined by immunohistochemistry or histology or by enzyme-linked immunosorbent assays, but these methods do not distinguish proMMPs from active MMPs. Therefore, these results are of limited value. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction
2
2007
... Many MMPs are expressed in the myocardium. In HCM, the expression of MMPs is altered, but not all MMPs are affected. Münch et al. found that MMP-1, MMP-2, MMP-3, and MMP-9 are involved in HCM [44]. They observed that increased serum MMP-2 levels in females were associated with lower fibrosis, while MMP-9 was positively associated with fibrosis in late gadolinium enhancement cardiac magnetic resonance (mean increase of 0.66 g/unit MMP-9 [0.50;0.82], p < 0.001), but neither serum MMP-1 nor MMP-3 levels were associated with cardiac fibrosis [44]. However, a cautionary note on these results is that the cardiac tissue concentration of MMPs was not evaluated, which might present a limitation. Regarding MMP-1 levels in systolic and diastolic HF, serum MMP-1 levels were increased in systolic HF patients, but not in diastolic HF patients [45, 46]. This result reminds us that MMP activity also fluctuates because heart remodeling during HF is a dynamic process. In diabetic mice (DM), myocardial fibrosis is clearly related to the expression of MMP7, MMP11, MMP13, and MMP16 in myocardial tissue [47]. Compared with the control group, levels of MMP7, MMP11, MMP13, and MMP16 were markedly higher, but levels of MMP2 were markedly lower in the DM group [47]. It is also important to note the measurement method of MMPs in all the above studies. The levels of different MMPs are determined by immunohistochemistry or histology or by enzyme-linked immunosorbent assays, but these methods do not distinguish proMMPs from active MMPs. Therefore, these results are of limited value. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Filling pressures and collagen metabolism in hypertensive patients with heart failure and normal ejection fraction
2
2010
... Many MMPs are expressed in the myocardium. In HCM, the expression of MMPs is altered, but not all MMPs are affected. Münch et al. found that MMP-1, MMP-2, MMP-3, and MMP-9 are involved in HCM [44]. They observed that increased serum MMP-2 levels in females were associated with lower fibrosis, while MMP-9 was positively associated with fibrosis in late gadolinium enhancement cardiac magnetic resonance (mean increase of 0.66 g/unit MMP-9 [0.50;0.82], p < 0.001), but neither serum MMP-1 nor MMP-3 levels were associated with cardiac fibrosis [44]. However, a cautionary note on these results is that the cardiac tissue concentration of MMPs was not evaluated, which might present a limitation. Regarding MMP-1 levels in systolic and diastolic HF, serum MMP-1 levels were increased in systolic HF patients, but not in diastolic HF patients [45, 46]. This result reminds us that MMP activity also fluctuates because heart remodeling during HF is a dynamic process. In diabetic mice (DM), myocardial fibrosis is clearly related to the expression of MMP7, MMP11, MMP13, and MMP16 in myocardial tissue [47]. Compared with the control group, levels of MMP7, MMP11, MMP13, and MMP16 were markedly higher, but levels of MMP2 were markedly lower in the DM group [47]. It is also important to note the measurement method of MMPs in all the above studies. The levels of different MMPs are determined by immunohistochemistry or histology or by enzyme-linked immunosorbent assays, but these methods do not distinguish proMMPs from active MMPs. Therefore, these results are of limited value. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Effects of hydrogen sulfide on myocardial fibrosis in diabetic rats: Changes in matrix metalloproteinases parameters
4
2015
... Many MMPs are expressed in the myocardium. In HCM, the expression of MMPs is altered, but not all MMPs are affected. Münch et al. found that MMP-1, MMP-2, MMP-3, and MMP-9 are involved in HCM [44]. They observed that increased serum MMP-2 levels in females were associated with lower fibrosis, while MMP-9 was positively associated with fibrosis in late gadolinium enhancement cardiac magnetic resonance (mean increase of 0.66 g/unit MMP-9 [0.50;0.82], p < 0.001), but neither serum MMP-1 nor MMP-3 levels were associated with cardiac fibrosis [44]. However, a cautionary note on these results is that the cardiac tissue concentration of MMPs was not evaluated, which might present a limitation. Regarding MMP-1 levels in systolic and diastolic HF, serum MMP-1 levels were increased in systolic HF patients, but not in diastolic HF patients [45, 46]. This result reminds us that MMP activity also fluctuates because heart remodeling during HF is a dynamic process. In diabetic mice (DM), myocardial fibrosis is clearly related to the expression of MMP7, MMP11, MMP13, and MMP16 in myocardial tissue [47]. Compared with the control group, levels of MMP7, MMP11, MMP13, and MMP16 were markedly higher, but levels of MMP2 were markedly lower in the DM group [47]. It is also important to note the measurement method of MMPs in all the above studies. The levels of different MMPs are determined by immunohistochemistry or histology or by enzyme-linked immunosorbent assays, but these methods do not distinguish proMMPs from active MMPs. Therefore, these results are of limited value. ...
... ]. Compared with the control group, levels of MMP7, MMP11, MMP13, and MMP16 were markedly higher, but levels of MMP2 were markedly lower in the DM group [47]. It is also important to note the measurement method of MMPs in all the above studies. The levels of different MMPs are determined by immunohistochemistry or histology or by enzyme-linked immunosorbent assays, but these methods do not distinguish proMMPs from active MMPs. Therefore, these results are of limited value. ...
... Tissue inhibitors of metalloproteinase (TIMPs) are a group of low molecular weight glycoproteins. TIMPs are produced and secreted by fibroblasts, epithelial cells, and endothelial cells, and are distributed among tissues and humors [48]. TIMPs can promote the differentiation of fibroblasts into myofibroblasts at the site of tissue injury [49]. In addition, TIMPs control the proteolytic activity of MMPs [50]. Specifically, TIMPs can specifically bind to the zinc ions in the catalytic site of MMPs through the cysteine residues, thus disbanding activated MMPs, or preventing inactive MMPs from becoming activated [47]. Therefore, the balance between MMP and TIMP expression is essential for reconstruction of the extracellular matrix in myocardial tissues [51]. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
The role of TIMPs in regulation of extracellular matrix proteolysis
1
2015
... Tissue inhibitors of metalloproteinase (TIMPs) are a group of low molecular weight glycoproteins. TIMPs are produced and secreted by fibroblasts, epithelial cells, and endothelial cells, and are distributed among tissues and humors [48]. TIMPs can promote the differentiation of fibroblasts into myofibroblasts at the site of tissue injury [49]. In addition, TIMPs control the proteolytic activity of MMPs [50]. Specifically, TIMPs can specifically bind to the zinc ions in the catalytic site of MMPs through the cysteine residues, thus disbanding activated MMPs, or preventing inactive MMPs from becoming activated [47]. Therefore, the balance between MMP and TIMP expression is essential for reconstruction of the extracellular matrix in myocardial tissues [51]. ...
Heterogeneous effects of tissue inhibitors of matrix metalloproteinases on cardiac fibroblasts
1
2005
... Tissue inhibitors of metalloproteinase (TIMPs) are a group of low molecular weight glycoproteins. TIMPs are produced and secreted by fibroblasts, epithelial cells, and endothelial cells, and are distributed among tissues and humors [48]. TIMPs can promote the differentiation of fibroblasts into myofibroblasts at the site of tissue injury [49]. In addition, TIMPs control the proteolytic activity of MMPs [50]. Specifically, TIMPs can specifically bind to the zinc ions in the catalytic site of MMPs through the cysteine residues, thus disbanding activated MMPs, or preventing inactive MMPs from becoming activated [47]. Therefore, the balance between MMP and TIMP expression is essential for reconstruction of the extracellular matrix in myocardial tissues [51]. ...
Tissue inhibitor of metalloproteinases (TIMP, aka EPA): structure, control of expression and biological functions
1
1993
... Tissue inhibitors of metalloproteinase (TIMPs) are a group of low molecular weight glycoproteins. TIMPs are produced and secreted by fibroblasts, epithelial cells, and endothelial cells, and are distributed among tissues and humors [48]. TIMPs can promote the differentiation of fibroblasts into myofibroblasts at the site of tissue injury [49]. In addition, TIMPs control the proteolytic activity of MMPs [50]. Specifically, TIMPs can specifically bind to the zinc ions in the catalytic site of MMPs through the cysteine residues, thus disbanding activated MMPs, or preventing inactive MMPs from becoming activated [47]. Therefore, the balance between MMP and TIMP expression is essential for reconstruction of the extracellular matrix in myocardial tissues [51]. ...
Regulation of MMP/TIMP by HUVEC transplantation attenuates ventricular remodeling in response to myocardial infarction
1
2014
... Tissue inhibitors of metalloproteinase (TIMPs) are a group of low molecular weight glycoproteins. TIMPs are produced and secreted by fibroblasts, epithelial cells, and endothelial cells, and are distributed among tissues and humors [48]. TIMPs can promote the differentiation of fibroblasts into myofibroblasts at the site of tissue injury [49]. In addition, TIMPs control the proteolytic activity of MMPs [50]. Specifically, TIMPs can specifically bind to the zinc ions in the catalytic site of MMPs through the cysteine residues, thus disbanding activated MMPs, or preventing inactive MMPs from becoming activated [47]. Therefore, the balance between MMP and TIMP expression is essential for reconstruction of the extracellular matrix in myocardial tissues [51]. ...
Tissue Inhibitor of Matrix Metalloproteinase-1 Promotes Myocardial Fibrosis by Mediating CD63-Integrin β1 Interaction
1
2017
... Currently, 4 types of TIMPs have been found to be associated with myocardial fibrosis. TIMP1 mediates the interaction between fibroblast membrane protein CD63 and integrin β1, which is widely expressed in various cells, thus triggering fibrosis [52-55]. At the cellular level, hirudin reverses Ang II-induced fibrosis by elevating TIMP-2 expression [56]. TIMP3 is the only TIMP that can inhibit the activity of TNF-α-converting enzyme and may have a protective effect against fibrosis by upregulating cytokines involved in myofibroblast activation and immunity [57]. In addition, Rizzi et al. investigated whether there was a correlation between cardiac TIMP-4 levels and cardiac hypertrophy in 2K1C hypertension rats [58]. They observed higher TIMP-4 levels (325 ± 68%) in hypertensive rats than in sham-operated animals at 75 days after 2K1C surgery. This result suggested that the increase in TIMP-4 activity was associated with concomitant development of cardiac hypertrophy. Another way to determine the severity of fibrosis is by measuring the MMP/TIMP ratio. Parkkonen et al. observed a large increase in serum TIMP-1 and MMP-2 levels in patients with takotsubo cardiomyopathy (TTC) compared with that in healthy controls, and proposed that the low MMP-8/TIMP-1 molar ratio may reflect increased transient fibrosis and decreased proteolysis [59]. This result indicates that the TIMP/MMP balance may play an important role in the development of myocardial fibrosis. In addition, the predicted value of TIMP-1 is also noteworthy. Frantz et al. investigated the association between TIMP-1 plasma levels and clinical endpoints (death due to any cause), and found that plasma TIMP-1 levels increased in HF patients as compared with healthy controls (1640 vs 735 ng/mL, respectively,) [60]. Moreover, the researchers also observed that patients with high TIMP-1 levels (1917 ng/mL) had a significantly higher mortality rate than patients with lower levels (1390 ng/mL). These results suggest that an increase in TIMP-1 indicates a poor prognosis in HF patients. ...
Integrin β1 is an essential factor in vasculogenic mimicry of human cancer cells
0
2018
Integrin β1 Optimizes Diabetogenic T Cell Migration and Function in the Pancreas
0
2018
Endothelial destabilization by angiopoietin-2 via integrin β1 activation
1
2015
... Currently, 4 types of TIMPs have been found to be associated with myocardial fibrosis. TIMP1 mediates the interaction between fibroblast membrane protein CD63 and integrin β1, which is widely expressed in various cells, thus triggering fibrosis [52-55]. At the cellular level, hirudin reverses Ang II-induced fibrosis by elevating TIMP-2 expression [56]. TIMP3 is the only TIMP that can inhibit the activity of TNF-α-converting enzyme and may have a protective effect against fibrosis by upregulating cytokines involved in myofibroblast activation and immunity [57]. In addition, Rizzi et al. investigated whether there was a correlation between cardiac TIMP-4 levels and cardiac hypertrophy in 2K1C hypertension rats [58]. They observed higher TIMP-4 levels (325 ± 68%) in hypertensive rats than in sham-operated animals at 75 days after 2K1C surgery. This result suggested that the increase in TIMP-4 activity was associated with concomitant development of cardiac hypertrophy. Another way to determine the severity of fibrosis is by measuring the MMP/TIMP ratio. Parkkonen et al. observed a large increase in serum TIMP-1 and MMP-2 levels in patients with takotsubo cardiomyopathy (TTC) compared with that in healthy controls, and proposed that the low MMP-8/TIMP-1 molar ratio may reflect increased transient fibrosis and decreased proteolysis [59]. This result indicates that the TIMP/MMP balance may play an important role in the development of myocardial fibrosis. In addition, the predicted value of TIMP-1 is also noteworthy. Frantz et al. investigated the association between TIMP-1 plasma levels and clinical endpoints (death due to any cause), and found that plasma TIMP-1 levels increased in HF patients as compared with healthy controls (1640 vs 735 ng/mL, respectively,) [60]. Moreover, the researchers also observed that patients with high TIMP-1 levels (1917 ng/mL) had a significantly higher mortality rate than patients with lower levels (1390 ng/mL). These results suggest that an increase in TIMP-1 indicates a poor prognosis in HF patients. ...
Hirudin Protects Ang II-Induced Myocardial Fibroblasts Fibrosis by Inhibiting the Extracellular Signal-Regulated Kinase1/2 (ERK1/2) Pathway
1
2018
... Currently, 4 types of TIMPs have been found to be associated with myocardial fibrosis. TIMP1 mediates the interaction between fibroblast membrane protein CD63 and integrin β1, which is widely expressed in various cells, thus triggering fibrosis [52-55]. At the cellular level, hirudin reverses Ang II-induced fibrosis by elevating TIMP-2 expression [56]. TIMP3 is the only TIMP that can inhibit the activity of TNF-α-converting enzyme and may have a protective effect against fibrosis by upregulating cytokines involved in myofibroblast activation and immunity [57]. In addition, Rizzi et al. investigated whether there was a correlation between cardiac TIMP-4 levels and cardiac hypertrophy in 2K1C hypertension rats [58]. They observed higher TIMP-4 levels (325 ± 68%) in hypertensive rats than in sham-operated animals at 75 days after 2K1C surgery. This result suggested that the increase in TIMP-4 activity was associated with concomitant development of cardiac hypertrophy. Another way to determine the severity of fibrosis is by measuring the MMP/TIMP ratio. Parkkonen et al. observed a large increase in serum TIMP-1 and MMP-2 levels in patients with takotsubo cardiomyopathy (TTC) compared with that in healthy controls, and proposed that the low MMP-8/TIMP-1 molar ratio may reflect increased transient fibrosis and decreased proteolysis [59]. This result indicates that the TIMP/MMP balance may play an important role in the development of myocardial fibrosis. In addition, the predicted value of TIMP-1 is also noteworthy. Frantz et al. investigated the association between TIMP-1 plasma levels and clinical endpoints (death due to any cause), and found that plasma TIMP-1 levels increased in HF patients as compared with healthy controls (1640 vs 735 ng/mL, respectively,) [60]. Moreover, the researchers also observed that patients with high TIMP-1 levels (1917 ng/mL) had a significantly higher mortality rate than patients with lower levels (1390 ng/mL). These results suggest that an increase in TIMP-1 indicates a poor prognosis in HF patients. ...
Fibrosis or hypertrophy: let TIMPs decide
1
2014
... Currently, 4 types of TIMPs have been found to be associated with myocardial fibrosis. TIMP1 mediates the interaction between fibroblast membrane protein CD63 and integrin β1, which is widely expressed in various cells, thus triggering fibrosis [52-55]. At the cellular level, hirudin reverses Ang II-induced fibrosis by elevating TIMP-2 expression [56]. TIMP3 is the only TIMP that can inhibit the activity of TNF-α-converting enzyme and may have a protective effect against fibrosis by upregulating cytokines involved in myofibroblast activation and immunity [57]. In addition, Rizzi et al. investigated whether there was a correlation between cardiac TIMP-4 levels and cardiac hypertrophy in 2K1C hypertension rats [58]. They observed higher TIMP-4 levels (325 ± 68%) in hypertensive rats than in sham-operated animals at 75 days after 2K1C surgery. This result suggested that the increase in TIMP-4 activity was associated with concomitant development of cardiac hypertrophy. Another way to determine the severity of fibrosis is by measuring the MMP/TIMP ratio. Parkkonen et al. observed a large increase in serum TIMP-1 and MMP-2 levels in patients with takotsubo cardiomyopathy (TTC) compared with that in healthy controls, and proposed that the low MMP-8/TIMP-1 molar ratio may reflect increased transient fibrosis and decreased proteolysis [59]. This result indicates that the TIMP/MMP balance may play an important role in the development of myocardial fibrosis. In addition, the predicted value of TIMP-1 is also noteworthy. Frantz et al. investigated the association between TIMP-1 plasma levels and clinical endpoints (death due to any cause), and found that plasma TIMP-1 levels increased in HF patients as compared with healthy controls (1640 vs 735 ng/mL, respectively,) [60]. Moreover, the researchers also observed that patients with high TIMP-1 levels (1917 ng/mL) had a significantly higher mortality rate than patients with lower levels (1390 ng/mL). These results suggest that an increase in TIMP-1 indicates a poor prognosis in HF patients. ...
Temporal changes in cardiac matrix metalloproteinase activity, oxidative stress, and TGF-β in renovascular hypertension-induced cardiac hypertrophy
2
2013
... Currently, 4 types of TIMPs have been found to be associated with myocardial fibrosis. TIMP1 mediates the interaction between fibroblast membrane protein CD63 and integrin β1, which is widely expressed in various cells, thus triggering fibrosis [52-55]. At the cellular level, hirudin reverses Ang II-induced fibrosis by elevating TIMP-2 expression [56]. TIMP3 is the only TIMP that can inhibit the activity of TNF-α-converting enzyme and may have a protective effect against fibrosis by upregulating cytokines involved in myofibroblast activation and immunity [57]. In addition, Rizzi et al. investigated whether there was a correlation between cardiac TIMP-4 levels and cardiac hypertrophy in 2K1C hypertension rats [58]. They observed higher TIMP-4 levels (325 ± 68%) in hypertensive rats than in sham-operated animals at 75 days after 2K1C surgery. This result suggested that the increase in TIMP-4 activity was associated with concomitant development of cardiac hypertrophy. Another way to determine the severity of fibrosis is by measuring the MMP/TIMP ratio. Parkkonen et al. observed a large increase in serum TIMP-1 and MMP-2 levels in patients with takotsubo cardiomyopathy (TTC) compared with that in healthy controls, and proposed that the low MMP-8/TIMP-1 molar ratio may reflect increased transient fibrosis and decreased proteolysis [59]. This result indicates that the TIMP/MMP balance may play an important role in the development of myocardial fibrosis. In addition, the predicted value of TIMP-1 is also noteworthy. Frantz et al. investigated the association between TIMP-1 plasma levels and clinical endpoints (death due to any cause), and found that plasma TIMP-1 levels increased in HF patients as compared with healthy controls (1640 vs 735 ng/mL, respectively,) [60]. Moreover, the researchers also observed that patients with high TIMP-1 levels (1917 ng/mL) had a significantly higher mortality rate than patients with lower levels (1390 ng/mL). These results suggest that an increase in TIMP-1 indicates a poor prognosis in HF patients. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Low MMP-8/TIMP-1 reflects left ventricle impairment in takotsubo cardiomyopathy and high TIMP-1 may help to differentiate it from acute coronary syndrome
2
2017
... Currently, 4 types of TIMPs have been found to be associated with myocardial fibrosis. TIMP1 mediates the interaction between fibroblast membrane protein CD63 and integrin β1, which is widely expressed in various cells, thus triggering fibrosis [52-55]. At the cellular level, hirudin reverses Ang II-induced fibrosis by elevating TIMP-2 expression [56]. TIMP3 is the only TIMP that can inhibit the activity of TNF-α-converting enzyme and may have a protective effect against fibrosis by upregulating cytokines involved in myofibroblast activation and immunity [57]. In addition, Rizzi et al. investigated whether there was a correlation between cardiac TIMP-4 levels and cardiac hypertrophy in 2K1C hypertension rats [58]. They observed higher TIMP-4 levels (325 ± 68%) in hypertensive rats than in sham-operated animals at 75 days after 2K1C surgery. This result suggested that the increase in TIMP-4 activity was associated with concomitant development of cardiac hypertrophy. Another way to determine the severity of fibrosis is by measuring the MMP/TIMP ratio. Parkkonen et al. observed a large increase in serum TIMP-1 and MMP-2 levels in patients with takotsubo cardiomyopathy (TTC) compared with that in healthy controls, and proposed that the low MMP-8/TIMP-1 molar ratio may reflect increased transient fibrosis and decreased proteolysis [59]. This result indicates that the TIMP/MMP balance may play an important role in the development of myocardial fibrosis. In addition, the predicted value of TIMP-1 is also noteworthy. Frantz et al. investigated the association between TIMP-1 plasma levels and clinical endpoints (death due to any cause), and found that plasma TIMP-1 levels increased in HF patients as compared with healthy controls (1640 vs 735 ng/mL, respectively,) [60]. Moreover, the researchers also observed that patients with high TIMP-1 levels (1917 ng/mL) had a significantly higher mortality rate than patients with lower levels (1390 ng/mL). These results suggest that an increase in TIMP-1 indicates a poor prognosis in HF patients. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Tissue inhibitor of metalloproteinases levels in patients with chronic heart failure: an independent predictor of mortality
2
2008
... Currently, 4 types of TIMPs have been found to be associated with myocardial fibrosis. TIMP1 mediates the interaction between fibroblast membrane protein CD63 and integrin β1, which is widely expressed in various cells, thus triggering fibrosis [52-55]. At the cellular level, hirudin reverses Ang II-induced fibrosis by elevating TIMP-2 expression [56]. TIMP3 is the only TIMP that can inhibit the activity of TNF-α-converting enzyme and may have a protective effect against fibrosis by upregulating cytokines involved in myofibroblast activation and immunity [57]. In addition, Rizzi et al. investigated whether there was a correlation between cardiac TIMP-4 levels and cardiac hypertrophy in 2K1C hypertension rats [58]. They observed higher TIMP-4 levels (325 ± 68%) in hypertensive rats than in sham-operated animals at 75 days after 2K1C surgery. This result suggested that the increase in TIMP-4 activity was associated with concomitant development of cardiac hypertrophy. Another way to determine the severity of fibrosis is by measuring the MMP/TIMP ratio. Parkkonen et al. observed a large increase in serum TIMP-1 and MMP-2 levels in patients with takotsubo cardiomyopathy (TTC) compared with that in healthy controls, and proposed that the low MMP-8/TIMP-1 molar ratio may reflect increased transient fibrosis and decreased proteolysis [59]. This result indicates that the TIMP/MMP balance may play an important role in the development of myocardial fibrosis. In addition, the predicted value of TIMP-1 is also noteworthy. Frantz et al. investigated the association between TIMP-1 plasma levels and clinical endpoints (death due to any cause), and found that plasma TIMP-1 levels increased in HF patients as compared with healthy controls (1640 vs 735 ng/mL, respectively,) [60]. Moreover, the researchers also observed that patients with high TIMP-1 levels (1917 ng/mL) had a significantly higher mortality rate than patients with lower levels (1390 ng/mL). These results suggest that an increase in TIMP-1 indicates a poor prognosis in HF patients. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Cardiac Fibrosis: The Fibroblast Awakens
1
2016
... Transforming growth factor-β (TGF-β) is a crucial profibrotic cytokine involved in myocardial fibrosis [61]. It is involved in the regulation of fibroblast proliferation, transformation, and migration, and production of the extracellular matrix [62]. As a cytokine upstream of LOX in cardiac fibroblasts, TGF-β activates fibrosis via the Smad3, PI3K/Akt, and MAPK signaling pathways [63]. In addition, the heterodimeric cell surface receptor complex composed of type I TGF-β receptors and type II TGF-β receptors plays an important role in signal transduction of cardiac fibroblasts [64, 65]. ...
Crosstalk between the TGF-β and WNT signalling pathways during cardiac fibrogenesis
1
2018
... Transforming growth factor-β (TGF-β) is a crucial profibrotic cytokine involved in myocardial fibrosis [61]. It is involved in the regulation of fibroblast proliferation, transformation, and migration, and production of the extracellular matrix [62]. As a cytokine upstream of LOX in cardiac fibroblasts, TGF-β activates fibrosis via the Smad3, PI3K/Akt, and MAPK signaling pathways [63]. In addition, the heterodimeric cell surface receptor complex composed of type I TGF-β receptors and type II TGF-β receptors plays an important role in signal transduction of cardiac fibroblasts [64, 65]. ...
Induction of cardiac fibroblast lysyl oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling
1
2011
... Transforming growth factor-β (TGF-β) is a crucial profibrotic cytokine involved in myocardial fibrosis [61]. It is involved in the regulation of fibroblast proliferation, transformation, and migration, and production of the extracellular matrix [62]. As a cytokine upstream of LOX in cardiac fibroblasts, TGF-β activates fibrosis via the Smad3, PI3K/Akt, and MAPK signaling pathways [63]. In addition, the heterodimeric cell surface receptor complex composed of type I TGF-β receptors and type II TGF-β receptors plays an important role in signal transduction of cardiac fibroblasts [64, 65]. ...
The role of TGF-beta signaling in myocardial infarction and cardiac remodeling
1
2007
... Transforming growth factor-β (TGF-β) is a crucial profibrotic cytokine involved in myocardial fibrosis [61]. It is involved in the regulation of fibroblast proliferation, transformation, and migration, and production of the extracellular matrix [62]. As a cytokine upstream of LOX in cardiac fibroblasts, TGF-β activates fibrosis via the Smad3, PI3K/Akt, and MAPK signaling pathways [63]. In addition, the heterodimeric cell surface receptor complex composed of type I TGF-β receptors and type II TGF-β receptors plays an important role in signal transduction of cardiac fibroblasts [64, 65]. ...
Myofibroblasts: trust your heart and let fate decide
1
2014
... Transforming growth factor-β (TGF-β) is a crucial profibrotic cytokine involved in myocardial fibrosis [61]. It is involved in the regulation of fibroblast proliferation, transformation, and migration, and production of the extracellular matrix [62]. As a cytokine upstream of LOX in cardiac fibroblasts, TGF-β activates fibrosis via the Smad3, PI3K/Akt, and MAPK signaling pathways [63]. In addition, the heterodimeric cell surface receptor complex composed of type I TGF-β receptors and type II TGF-β receptors plays an important role in signal transduction of cardiac fibroblasts [64, 65]. ...
Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction
2
2008
... In various animal models of heart disease, inhibition of the effect of TGF-β successfully reduced or prevented the development of fibrosis. In MI mice, anti-TGF-β treatment after coronary artery ligation increased the expression of MMPs and decreased the production of collagen [66]. Similarly, in pressure-overloaded rats, the delivery of anti-TGF-β neutralizing antibody inhibited the activation of fibroblasts and prevented the induction of collagen type I and type III mRNA [67]. These observations indicated that anti-TGF treatment prevented excessive extracellular matrix deposition. Moreover, Koitabashi et al. observed that deletion of the TGF-β type II receptor (TβR2) gene in cardiomyocytes inhibited cardiac hypertrophy, and that transverse aortic constriction (TAC) mice receiving anti-TGF agents displayed significantly suppressed perivascular and interstitial fibrosis [68]. Taken together, these data emphasize the key role of TGF-β in cardiac fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats
2
2002
... In various animal models of heart disease, inhibition of the effect of TGF-β successfully reduced or prevented the development of fibrosis. In MI mice, anti-TGF-β treatment after coronary artery ligation increased the expression of MMPs and decreased the production of collagen [66]. Similarly, in pressure-overloaded rats, the delivery of anti-TGF-β neutralizing antibody inhibited the activation of fibroblasts and prevented the induction of collagen type I and type III mRNA [67]. These observations indicated that anti-TGF treatment prevented excessive extracellular matrix deposition. Moreover, Koitabashi et al. observed that deletion of the TGF-β type II receptor (TβR2) gene in cardiomyocytes inhibited cardiac hypertrophy, and that transverse aortic constriction (TAC) mice receiving anti-TGF agents displayed significantly suppressed perivascular and interstitial fibrosis [68]. Taken together, these data emphasize the key role of TGF-β in cardiac fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload
2
2011
... In various animal models of heart disease, inhibition of the effect of TGF-β successfully reduced or prevented the development of fibrosis. In MI mice, anti-TGF-β treatment after coronary artery ligation increased the expression of MMPs and decreased the production of collagen [66]. Similarly, in pressure-overloaded rats, the delivery of anti-TGF-β neutralizing antibody inhibited the activation of fibroblasts and prevented the induction of collagen type I and type III mRNA [67]. These observations indicated that anti-TGF treatment prevented excessive extracellular matrix deposition. Moreover, Koitabashi et al. observed that deletion of the TGF-β type II receptor (TβR2) gene in cardiomyocytes inhibited cardiac hypertrophy, and that transverse aortic constriction (TAC) mice receiving anti-TGF agents displayed significantly suppressed perivascular and interstitial fibrosis [68]. Taken together, these data emphasize the key role of TGF-β in cardiac fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Oxymatrine Inhibits Renal Tubular EMT Induced by High Glucose via Upregulation of SnoN and Inhibition of TGF-β1/Smad Signaling Pathway
1
2016
... Smads, as key intracellular effectors of the TGF-β1 class, and play a central role in cardiac remodeling [69, 70]. Research has suggested that the up-regulatory effect of TGF-β3 on the migration, proliferation, and collagen synthesis of human cardiac fibroblasts may be attributed to the action of Smad7 [71]. Khalil et al. found that the absence of Smad3, Smad2/3, or Tgfbr1/2 markedly attenuated cardiac fibrosis after 12 weeks of TAC stimulation in rats [72]. In Smad3-deficient mice, the collagen content in the infarcted hearts decreased at 7 days after reperfusion, and collagen deposition in the uninfarcted, remodeling myocardium was markedly attenuated [73]. These findings suggest that Smad gene disruption leads to attenuation of the remodeling process. ...
TGF-β1/Smad signaling pathway regulates epithelial-to-mesenchymal transition in esophageal squamous cell carcinoma: in vitro and clinical analyses of cell lines and nomadic Kazakh patients from northwest Xinjiang, China
1
2014
... Smads, as key intracellular effectors of the TGF-β1 class, and play a central role in cardiac remodeling [69, 70]. Research has suggested that the up-regulatory effect of TGF-β3 on the migration, proliferation, and collagen synthesis of human cardiac fibroblasts may be attributed to the action of Smad7 [71]. Khalil et al. found that the absence of Smad3, Smad2/3, or Tgfbr1/2 markedly attenuated cardiac fibrosis after 12 weeks of TAC stimulation in rats [72]. In Smad3-deficient mice, the collagen content in the infarcted hearts decreased at 7 days after reperfusion, and collagen deposition in the uninfarcted, remodeling myocardium was markedly attenuated [73]. These findings suggest that Smad gene disruption leads to attenuation of the remodeling process. ...
The role and mechanism of transforming growth factor beta 3 in human myocardial infarction-induced myocardial fibrosis
1
2019
... Smads, as key intracellular effectors of the TGF-β1 class, and play a central role in cardiac remodeling [69, 70]. Research has suggested that the up-regulatory effect of TGF-β3 on the migration, proliferation, and collagen synthesis of human cardiac fibroblasts may be attributed to the action of Smad7 [71]. Khalil et al. found that the absence of Smad3, Smad2/3, or Tgfbr1/2 markedly attenuated cardiac fibrosis after 12 weeks of TAC stimulation in rats [72]. In Smad3-deficient mice, the collagen content in the infarcted hearts decreased at 7 days after reperfusion, and collagen deposition in the uninfarcted, remodeling myocardium was markedly attenuated [73]. These findings suggest that Smad gene disruption leads to attenuation of the remodeling process. ...
... Smads, as key intracellular effectors of the TGF-β1 class, and play a central role in cardiac remodeling [69, 70]. Research has suggested that the up-regulatory effect of TGF-β3 on the migration, proliferation, and collagen synthesis of human cardiac fibroblasts may be attributed to the action of Smad7 [71]. Khalil et al. found that the absence of Smad3, Smad2/3, or Tgfbr1/2 markedly attenuated cardiac fibrosis after 12 weeks of TAC stimulation in rats [72]. In Smad3-deficient mice, the collagen content in the infarcted hearts decreased at 7 days after reperfusion, and collagen deposition in the uninfarcted, remodeling myocardium was markedly attenuated [73]. These findings suggest that Smad gene disruption leads to attenuation of the remodeling process. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling
2
2007
... Smads, as key intracellular effectors of the TGF-β1 class, and play a central role in cardiac remodeling [69, 70]. Research has suggested that the up-regulatory effect of TGF-β3 on the migration, proliferation, and collagen synthesis of human cardiac fibroblasts may be attributed to the action of Smad7 [71]. Khalil et al. found that the absence of Smad3, Smad2/3, or Tgfbr1/2 markedly attenuated cardiac fibrosis after 12 weeks of TAC stimulation in rats [72]. In Smad3-deficient mice, the collagen content in the infarcted hearts decreased at 7 days after reperfusion, and collagen deposition in the uninfarcted, remodeling myocardium was markedly attenuated [73]. These findings suggest that Smad gene disruption leads to attenuation of the remodeling process. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
The modular architecture of a new family of growth regulators related to connective tissue growth factor
1
1993
... Connective tissue growth factor (CTGF) belongs to the CCN family of multifunctional matricellular proteins, and it is involved in the process of triggering fibrosis in multiple organs and tissues, including the heart [74, 75]. Many in vitro studies have demonstrated that CTGF promotes the differentiation of fibroblasts into myofibroblasts and enhances extracellular matrix production [76-78]. CTGF is also a critical mediator of the downstream signaling of the profibrotic cytokine TGF-β [79, 80]. In DCM model rats, CTGF/CCN2 gene silencing improved cardiac function, attenuated myocardial fibrosis, and left ventricular hypertrophy [81]. In an in vitro study, β-adrenergic receptor overstimulation could induce the synthesis and secretion of CTGF in cardiomyocytes, thereby affecting the activation of cardiac fibroblasts [82]. Dean et al. observed the expression of spatiotemporal CTGF during the development of myocardial fibrosis in experimental MI rats [83]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis. ...
Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis
1
2008
... Connective tissue growth factor (CTGF) belongs to the CCN family of multifunctional matricellular proteins, and it is involved in the process of triggering fibrosis in multiple organs and tissues, including the heart [74, 75]. Many in vitro studies have demonstrated that CTGF promotes the differentiation of fibroblasts into myofibroblasts and enhances extracellular matrix production [76-78]. CTGF is also a critical mediator of the downstream signaling of the profibrotic cytokine TGF-β [79, 80]. In DCM model rats, CTGF/CCN2 gene silencing improved cardiac function, attenuated myocardial fibrosis, and left ventricular hypertrophy [81]. In an in vitro study, β-adrenergic receptor overstimulation could induce the synthesis and secretion of CTGF in cardiomyocytes, thereby affecting the activation of cardiac fibroblasts [82]. Dean et al. observed the expression of spatiotemporal CTGF during the development of myocardial fibrosis in experimental MI rats [83]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis. ...
Mediation of transforming growth factor-beta(1)-stimulated matrix contraction by fibroblasts: a role for connective tissue growth factor in contractile scarring
1
2003
... Connective tissue growth factor (CTGF) belongs to the CCN family of multifunctional matricellular proteins, and it is involved in the process of triggering fibrosis in multiple organs and tissues, including the heart [74, 75]. Many in vitro studies have demonstrated that CTGF promotes the differentiation of fibroblasts into myofibroblasts and enhances extracellular matrix production [76-78]. CTGF is also a critical mediator of the downstream signaling of the profibrotic cytokine TGF-β [79, 80]. In DCM model rats, CTGF/CCN2 gene silencing improved cardiac function, attenuated myocardial fibrosis, and left ventricular hypertrophy [81]. In an in vitro study, β-adrenergic receptor overstimulation could induce the synthesis and secretion of CTGF in cardiomyocytes, thereby affecting the activation of cardiac fibroblasts [82]. Dean et al. observed the expression of spatiotemporal CTGF during the development of myocardial fibrosis in experimental MI rats [83]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis. ...
Combinatorial signaling pathways determine fibroblast proliferation and myofibroblast differentiation
0
2004
Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor
1
1996
... Connective tissue growth factor (CTGF) belongs to the CCN family of multifunctional matricellular proteins, and it is involved in the process of triggering fibrosis in multiple organs and tissues, including the heart [74, 75]. Many in vitro studies have demonstrated that CTGF promotes the differentiation of fibroblasts into myofibroblasts and enhances extracellular matrix production [76-78]. CTGF is also a critical mediator of the downstream signaling of the profibrotic cytokine TGF-β [79, 80]. In DCM model rats, CTGF/CCN2 gene silencing improved cardiac function, attenuated myocardial fibrosis, and left ventricular hypertrophy [81]. In an in vitro study, β-adrenergic receptor overstimulation could induce the synthesis and secretion of CTGF in cardiomyocytes, thereby affecting the activation of cardiac fibroblasts [82]. Dean et al. observed the expression of spatiotemporal CTGF during the development of myocardial fibrosis in experimental MI rats [83]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis. ...
Gene regulation of connective tissue growth factor: new targets for antifibrotic therapy?
1
2002
... Connective tissue growth factor (CTGF) belongs to the CCN family of multifunctional matricellular proteins, and it is involved in the process of triggering fibrosis in multiple organs and tissues, including the heart [74, 75]. Many in vitro studies have demonstrated that CTGF promotes the differentiation of fibroblasts into myofibroblasts and enhances extracellular matrix production [76-78]. CTGF is also a critical mediator of the downstream signaling of the profibrotic cytokine TGF-β [79, 80]. In DCM model rats, CTGF/CCN2 gene silencing improved cardiac function, attenuated myocardial fibrosis, and left ventricular hypertrophy [81]. In an in vitro study, β-adrenergic receptor overstimulation could induce the synthesis and secretion of CTGF in cardiomyocytes, thereby affecting the activation of cardiac fibroblasts [82]. Dean et al. observed the expression of spatiotemporal CTGF during the development of myocardial fibrosis in experimental MI rats [83]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis. ...
Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta
1
2002
... Connective tissue growth factor (CTGF) belongs to the CCN family of multifunctional matricellular proteins, and it is involved in the process of triggering fibrosis in multiple organs and tissues, including the heart [74, 75]. Many in vitro studies have demonstrated that CTGF promotes the differentiation of fibroblasts into myofibroblasts and enhances extracellular matrix production [76-78]. CTGF is also a critical mediator of the downstream signaling of the profibrotic cytokine TGF-β [79, 80]. In DCM model rats, CTGF/CCN2 gene silencing improved cardiac function, attenuated myocardial fibrosis, and left ventricular hypertrophy [81]. In an in vitro study, β-adrenergic receptor overstimulation could induce the synthesis and secretion of CTGF in cardiomyocytes, thereby affecting the activation of cardiac fibroblasts [82]. Dean et al. observed the expression of spatiotemporal CTGF during the development of myocardial fibrosis in experimental MI rats [83]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis. ...
Silencing CTGF/CCN2 inactivates the MAPK signaling pathway to alleviate myocardial fibrosis and left ventricular hypertrophy in rats with dilated cardiomyopathy
2
2018
... Connective tissue growth factor (CTGF) belongs to the CCN family of multifunctional matricellular proteins, and it is involved in the process of triggering fibrosis in multiple organs and tissues, including the heart [74, 75]. Many in vitro studies have demonstrated that CTGF promotes the differentiation of fibroblasts into myofibroblasts and enhances extracellular matrix production [76-78]. CTGF is also a critical mediator of the downstream signaling of the profibrotic cytokine TGF-β [79, 80]. In DCM model rats, CTGF/CCN2 gene silencing improved cardiac function, attenuated myocardial fibrosis, and left ventricular hypertrophy [81]. In an in vitro study, β-adrenergic receptor overstimulation could induce the synthesis and secretion of CTGF in cardiomyocytes, thereby affecting the activation of cardiac fibroblasts [82]. Dean et al. observed the expression of spatiotemporal CTGF during the development of myocardial fibrosis in experimental MI rats [83]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Sustained β-AR stimulation induces synthesis and secretion of growth factors in cardiac myocytes that affect on cardiac fibroblast activation
1
2018
... Connective tissue growth factor (CTGF) belongs to the CCN family of multifunctional matricellular proteins, and it is involved in the process of triggering fibrosis in multiple organs and tissues, including the heart [74, 75]. Many in vitro studies have demonstrated that CTGF promotes the differentiation of fibroblasts into myofibroblasts and enhances extracellular matrix production [76-78]. CTGF is also a critical mediator of the downstream signaling of the profibrotic cytokine TGF-β [79, 80]. In DCM model rats, CTGF/CCN2 gene silencing improved cardiac function, attenuated myocardial fibrosis, and left ventricular hypertrophy [81]. In an in vitro study, β-adrenergic receptor overstimulation could induce the synthesis and secretion of CTGF in cardiomyocytes, thereby affecting the activation of cardiac fibroblasts [82]. Dean et al. observed the expression of spatiotemporal CTGF during the development of myocardial fibrosis in experimental MI rats [83]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis. ...
Connective tissue growth factor and cardiac fibrosis after myocardial infarction
3
2005
... Connective tissue growth factor (CTGF) belongs to the CCN family of multifunctional matricellular proteins, and it is involved in the process of triggering fibrosis in multiple organs and tissues, including the heart [74, 75]. Many in vitro studies have demonstrated that CTGF promotes the differentiation of fibroblasts into myofibroblasts and enhances extracellular matrix production [76-78]. CTGF is also a critical mediator of the downstream signaling of the profibrotic cytokine TGF-β [79, 80]. In DCM model rats, CTGF/CCN2 gene silencing improved cardiac function, attenuated myocardial fibrosis, and left ventricular hypertrophy [81]. In an in vitro study, β-adrenergic receptor overstimulation could induce the synthesis and secretion of CTGF in cardiomyocytes, thereby affecting the activation of cardiac fibroblasts [82]. Dean et al. observed the expression of spatiotemporal CTGF during the development of myocardial fibrosis in experimental MI rats [83]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis. ...
... ]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Plasma connective tissue growth factor is a novel potential biomarker of cardiac dysfunction in patients with chronic heart failure
2
2008
... Connective tissue growth factor (CTGF) belongs to the CCN family of multifunctional matricellular proteins, and it is involved in the process of triggering fibrosis in multiple organs and tissues, including the heart [74, 75]. Many in vitro studies have demonstrated that CTGF promotes the differentiation of fibroblasts into myofibroblasts and enhances extracellular matrix production [76-78]. CTGF is also a critical mediator of the downstream signaling of the profibrotic cytokine TGF-β [79, 80]. In DCM model rats, CTGF/CCN2 gene silencing improved cardiac function, attenuated myocardial fibrosis, and left ventricular hypertrophy [81]. In an in vitro study, β-adrenergic receptor overstimulation could induce the synthesis and secretion of CTGF in cardiomyocytes, thereby affecting the activation of cardiac fibroblasts [82]. Dean et al. observed the expression of spatiotemporal CTGF during the development of myocardial fibrosis in experimental MI rats [83]. They confirmed that the expression of CTGF protein and mRNA was upregulated rapidly in the infarcted region within a few days after MI [83]. These findings suggest that CTGF plays a key role in myocardial fibrosis. Furthermore, Koitabashi et al. proposed that plasma CTGF could be used as a diagnostic marker for chronic HF [84]. They found that plasma CTGF levels in symptomatic choric HF patients were significantly increased in proportion to their NYHA classes and were correlated with plasma BNP concentration (r = 0.395, P<0.01). However, they did not measure the concentration of CTGF in aortic root blood and coronary sinus blood; thus, the source of CTGF production is unknown. In brief, CTGF may be a predictor of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Plasma soluble corin in patients with heart failure
3
2010
... Corin is a transmembrane protease that is mainly expressed in cardiomyocytes [85]. It is closely correlated with natriuretic peptides that regulate many signaling functions, such as cGMP levels, vasodilation, natriuresis, fibrosis, etc. [86, 87]. In DCM model mice, increasing the expression of cardiac corin not only reduced fibrosis and HF, but also increased survival [88]. This finding indicates that corin correlates with the prognosis of DCM. In a cross-sectional study, plasma corin levels were lower in HF patients (365 pg/mL [±SD, 259 pg/mL]; P < 0.001) than in healthy controls, and this reduction was closely related to the severity of the disease (P < 0.001 for NYHA class II vs. class IV; P < 0.05 for NYHA class III vs. class IV) [85]. The results demonstrated that plasma corin may indicate pathological conditions in the heart. In canines with HF, the staining intensity of collagen was markedly elevated, while the expression of corin mRNA and protein was lower than that in the control group [89]. Contrary to these findings, Tran et al. observed upregulation of corin gene expression in the failing myocardium and in hypertrophic cardiomyocytes [90]. These studies suggest that the expression of corin may differ according to the disease state. In summary, corin may be an attractive diagnostic and prognostic biomarker of myocardial fibrosis. ...
... ]. This finding indicates that corin correlates with the prognosis of DCM. In a cross-sectional study, plasma corin levels were lower in HF patients (365 pg/mL [±SD, 259 pg/mL]; P < 0.001) than in healthy controls, and this reduction was closely related to the severity of the disease (P < 0.001 for NYHA class II vs. class IV; P < 0.05 for NYHA class III vs. class IV) [85]. The results demonstrated that plasma corin may indicate pathological conditions in the heart. In canines with HF, the staining intensity of collagen was markedly elevated, while the expression of corin mRNA and protein was lower than that in the control group [89]. Contrary to these findings, Tran et al. observed upregulation of corin gene expression in the failing myocardium and in hypertrophic cardiomyocytes [90]. These studies suggest that the expression of corin may differ according to the disease state. In summary, corin may be an attractive diagnostic and prognostic biomarker of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme
1
2000
... Corin is a transmembrane protease that is mainly expressed in cardiomyocytes [85]. It is closely correlated with natriuretic peptides that regulate many signaling functions, such as cGMP levels, vasodilation, natriuresis, fibrosis, etc. [86, 87]. In DCM model mice, increasing the expression of cardiac corin not only reduced fibrosis and HF, but also increased survival [88]. This finding indicates that corin correlates with the prognosis of DCM. In a cross-sectional study, plasma corin levels were lower in HF patients (365 pg/mL [±SD, 259 pg/mL]; P < 0.001) than in healthy controls, and this reduction was closely related to the severity of the disease (P < 0.001 for NYHA class II vs. class IV; P < 0.05 for NYHA class III vs. class IV) [85]. The results demonstrated that plasma corin may indicate pathological conditions in the heart. In canines with HF, the staining intensity of collagen was markedly elevated, while the expression of corin mRNA and protein was lower than that in the control group [89]. Contrary to these findings, Tran et al. observed upregulation of corin gene expression in the failing myocardium and in hypertrophic cardiomyocytes [90]. These studies suggest that the expression of corin may differ according to the disease state. In summary, corin may be an attractive diagnostic and prognostic biomarker of myocardial fibrosis. ...
Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions
1
2006
... Corin is a transmembrane protease that is mainly expressed in cardiomyocytes [85]. It is closely correlated with natriuretic peptides that regulate many signaling functions, such as cGMP levels, vasodilation, natriuresis, fibrosis, etc. [86, 87]. In DCM model mice, increasing the expression of cardiac corin not only reduced fibrosis and HF, but also increased survival [88]. This finding indicates that corin correlates with the prognosis of DCM. In a cross-sectional study, plasma corin levels were lower in HF patients (365 pg/mL [±SD, 259 pg/mL]; P < 0.001) than in healthy controls, and this reduction was closely related to the severity of the disease (P < 0.001 for NYHA class II vs. class IV; P < 0.05 for NYHA class III vs. class IV) [85]. The results demonstrated that plasma corin may indicate pathological conditions in the heart. In canines with HF, the staining intensity of collagen was markedly elevated, while the expression of corin mRNA and protein was lower than that in the control group [89]. Contrary to these findings, Tran et al. observed upregulation of corin gene expression in the failing myocardium and in hypertrophic cardiomyocytes [90]. These studies suggest that the expression of corin may differ according to the disease state. In summary, corin may be an attractive diagnostic and prognostic biomarker of myocardial fibrosis. ...
Corin overexpression improves cardiac function, heart failure, and survival in mice with dilated cardiomyopathy
1
2013
... Corin is a transmembrane protease that is mainly expressed in cardiomyocytes [85]. It is closely correlated with natriuretic peptides that regulate many signaling functions, such as cGMP levels, vasodilation, natriuresis, fibrosis, etc. [86, 87]. In DCM model mice, increasing the expression of cardiac corin not only reduced fibrosis and HF, but also increased survival [88]. This finding indicates that corin correlates with the prognosis of DCM. In a cross-sectional study, plasma corin levels were lower in HF patients (365 pg/mL [±SD, 259 pg/mL]; P < 0.001) than in healthy controls, and this reduction was closely related to the severity of the disease (P < 0.001 for NYHA class II vs. class IV; P < 0.05 for NYHA class III vs. class IV) [85]. The results demonstrated that plasma corin may indicate pathological conditions in the heart. In canines with HF, the staining intensity of collagen was markedly elevated, while the expression of corin mRNA and protein was lower than that in the control group [89]. Contrary to these findings, Tran et al. observed upregulation of corin gene expression in the failing myocardium and in hypertrophic cardiomyocytes [90]. These studies suggest that the expression of corin may differ according to the disease state. In summary, corin may be an attractive diagnostic and prognostic biomarker of myocardial fibrosis. ...
Differential expression of the pro-natriuretic peptide convertases corin and furin in experimental heart failure and atrial fibrosis
2
2013
... Corin is a transmembrane protease that is mainly expressed in cardiomyocytes [85]. It is closely correlated with natriuretic peptides that regulate many signaling functions, such as cGMP levels, vasodilation, natriuresis, fibrosis, etc. [86, 87]. In DCM model mice, increasing the expression of cardiac corin not only reduced fibrosis and HF, but also increased survival [88]. This finding indicates that corin correlates with the prognosis of DCM. In a cross-sectional study, plasma corin levels were lower in HF patients (365 pg/mL [±SD, 259 pg/mL]; P < 0.001) than in healthy controls, and this reduction was closely related to the severity of the disease (P < 0.001 for NYHA class II vs. class IV; P < 0.05 for NYHA class III vs. class IV) [85]. The results demonstrated that plasma corin may indicate pathological conditions in the heart. In canines with HF, the staining intensity of collagen was markedly elevated, while the expression of corin mRNA and protein was lower than that in the control group [89]. Contrary to these findings, Tran et al. observed upregulation of corin gene expression in the failing myocardium and in hypertrophic cardiomyocytes [90]. These studies suggest that the expression of corin may differ according to the disease state. In summary, corin may be an attractive diagnostic and prognostic biomarker of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Upregulation of corin gene expression in hypertrophic cardiomyocytes and failing myocardium
2
2004
... Corin is a transmembrane protease that is mainly expressed in cardiomyocytes [85]. It is closely correlated with natriuretic peptides that regulate many signaling functions, such as cGMP levels, vasodilation, natriuresis, fibrosis, etc. [86, 87]. In DCM model mice, increasing the expression of cardiac corin not only reduced fibrosis and HF, but also increased survival [88]. This finding indicates that corin correlates with the prognosis of DCM. In a cross-sectional study, plasma corin levels were lower in HF patients (365 pg/mL [±SD, 259 pg/mL]; P < 0.001) than in healthy controls, and this reduction was closely related to the severity of the disease (P < 0.001 for NYHA class II vs. class IV; P < 0.05 for NYHA class III vs. class IV) [85]. The results demonstrated that plasma corin may indicate pathological conditions in the heart. In canines with HF, the staining intensity of collagen was markedly elevated, while the expression of corin mRNA and protein was lower than that in the control group [89]. Contrary to these findings, Tran et al. observed upregulation of corin gene expression in the failing myocardium and in hypertrophic cardiomyocytes [90]. These studies suggest that the expression of corin may differ according to the disease state. In summary, corin may be an attractive diagnostic and prognostic biomarker of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders
1
2011
... Recently, endothelial-to-mesenchymal transition (EndoMT) has been considered as a potential mechanism in pathological fibrosis. In the complex biological process of EndoMT, endothelial cells lose their specific cellular markers, and then acquire a mesenchymal or myofibroblast phenotype to initiate expression of mesenchymal cell products, such as α-smooth muscle actin (α-SMA), vimentin, and fibronectin [91]. In a rat model of isoproterenol (ISO) -induced myocardial fibrosis, cardiac α-SMA and vimentin levels increased, peaking on day 3, and then gradually decreased [92]. This result suggests that α-SMA and vimentin activity fluctuates in the fibrosis process. α-SMA, fibronectin, and vimentin are also involved in the process of diabetic cardiomyopathy [93]. The immunohistochemistry data showed that diabetes enhanced the expression of cardiac α-SMA, fibronectin, and vimentin compared with normal rats. Moreover, in vitro treatment of mouse embryonic fibroblasts with WF-A reduced the stability of collagen mRNA, but in the absence of vimentin, WF-A did not change the half-life of collagen mRNAs [94]. We therefore conclude that vimentin filaments play a crucial role in collagen expression. In short, α-SMA, vimentin, and fibronectin might be essential elements in cardiac fibrosis. ...
Immunohistochemical characterization of myofibroblasts appearing in isoproterenol-induced rat myocardial fibrosis
2
2019
... Recently, endothelial-to-mesenchymal transition (EndoMT) has been considered as a potential mechanism in pathological fibrosis. In the complex biological process of EndoMT, endothelial cells lose their specific cellular markers, and then acquire a mesenchymal or myofibroblast phenotype to initiate expression of mesenchymal cell products, such as α-smooth muscle actin (α-SMA), vimentin, and fibronectin [91]. In a rat model of isoproterenol (ISO) -induced myocardial fibrosis, cardiac α-SMA and vimentin levels increased, peaking on day 3, and then gradually decreased [92]. This result suggests that α-SMA and vimentin activity fluctuates in the fibrosis process. α-SMA, fibronectin, and vimentin are also involved in the process of diabetic cardiomyopathy [93]. The immunohistochemistry data showed that diabetes enhanced the expression of cardiac α-SMA, fibronectin, and vimentin compared with normal rats. Moreover, in vitro treatment of mouse embryonic fibroblasts with WF-A reduced the stability of collagen mRNA, but in the absence of vimentin, WF-A did not change the half-life of collagen mRNAs [94]. We therefore conclude that vimentin filaments play a crucial role in collagen expression. In short, α-SMA, vimentin, and fibronectin might be essential elements in cardiac fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
MiR-18a-5p inhibits endothelial-mesenchymal transition and cardiac fibrosis through the Notch2 pathway
2
2017
... Recently, endothelial-to-mesenchymal transition (EndoMT) has been considered as a potential mechanism in pathological fibrosis. In the complex biological process of EndoMT, endothelial cells lose their specific cellular markers, and then acquire a mesenchymal or myofibroblast phenotype to initiate expression of mesenchymal cell products, such as α-smooth muscle actin (α-SMA), vimentin, and fibronectin [91]. In a rat model of isoproterenol (ISO) -induced myocardial fibrosis, cardiac α-SMA and vimentin levels increased, peaking on day 3, and then gradually decreased [92]. This result suggests that α-SMA and vimentin activity fluctuates in the fibrosis process. α-SMA, fibronectin, and vimentin are also involved in the process of diabetic cardiomyopathy [93]. The immunohistochemistry data showed that diabetes enhanced the expression of cardiac α-SMA, fibronectin, and vimentin compared with normal rats. Moreover, in vitro treatment of mouse embryonic fibroblasts with WF-A reduced the stability of collagen mRNA, but in the absence of vimentin, WF-A did not change the half-life of collagen mRNAs [94]. We therefore conclude that vimentin filaments play a crucial role in collagen expression. In short, α-SMA, vimentin, and fibronectin might be essential elements in cardiac fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Withaferin-A reduces type I collagen expression in vitro and inhibits development of myocardial fibrosis in vivo
2
2012
... Recently, endothelial-to-mesenchymal transition (EndoMT) has been considered as a potential mechanism in pathological fibrosis. In the complex biological process of EndoMT, endothelial cells lose their specific cellular markers, and then acquire a mesenchymal or myofibroblast phenotype to initiate expression of mesenchymal cell products, such as α-smooth muscle actin (α-SMA), vimentin, and fibronectin [91]. In a rat model of isoproterenol (ISO) -induced myocardial fibrosis, cardiac α-SMA and vimentin levels increased, peaking on day 3, and then gradually decreased [92]. This result suggests that α-SMA and vimentin activity fluctuates in the fibrosis process. α-SMA, fibronectin, and vimentin are also involved in the process of diabetic cardiomyopathy [93]. The immunohistochemistry data showed that diabetes enhanced the expression of cardiac α-SMA, fibronectin, and vimentin compared with normal rats. Moreover, in vitro treatment of mouse embryonic fibroblasts with WF-A reduced the stability of collagen mRNA, but in the absence of vimentin, WF-A did not change the half-life of collagen mRNAs [94]. We therefore conclude that vimentin filaments play a crucial role in collagen expression. In short, α-SMA, vimentin, and fibronectin might be essential elements in cardiac fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis
1
2013
... Galectin-3 (Gal-3), a β-galactoside-binding lectin, plays a regulatory role in fibrogenesis. It is widely expressed by various types of inflammatory cells, binds to extracellular glycoproteins, and activates fibroblasts to increase collagen I deposition [95-97]. Growing evidence indicates that high levels of circulating Gal-3 are associated with an increased risk of adverse cardiovascular events, such as HF, myocardial infarction, dilated cardiomyopathy, and fibrogenesis [98-101]. In mice with ischemia/reperfusion (I/R) injury, there was a two-fold increase in plasma Gal-3 levels, and in the ischemic myocardium, Gal-3 was upregulated seven-fold at the mRNA level and 30-fold at the protein level [102]. In addition, that study revealed that plasma Gal-3 concentrations were always higher in DCM/ICM patients with HF than in non-HF subjects, but no trans-cardiac or trans-hepatic concentration gradient of Gal-3 was found. The aforementioned studies were limited in that they did not obtain myocardial biopsies to measure cardiac Gal-3 expression. Moreover, serum Gal-3 is considered to be associated with inflammation and cardiovascular fibrosis. In patients with AMI, serum Gal-3 levels increased immediately after AMI and then declined significantly within 5 days [103]. These data indicate that Gal-3 is closely related to the formation, destabilization, and rupture of plaque. Besides observational studies of Gal-3 levels, several studies have investigated the predictive value of Gal-3. Van Kimmenade et al. observed that an increased circulating Gal-3 level was the best independent predictor of 60-day mortality (odds ratio 10.3, P < 0.01) in patients with acute HF [104]. In patients with coronary artery disease (CAD), Maiolino et al. found that patients with Gal-3 levels in the highest tertile were more prone to death due to cardiovascular causes (25.2%) than those with Gal-3 levels in the intermediate and lower tertiles (13.6% and 7.5%, respectively; P < 0.001), and the plasma Gal-3 cutoff value for cardiovascular death prediction was 27.7 ng/mL [105]. Unfortunately, the loss of 25% of the patients to follow-up may have limited the results of the study. Overall, these findings confirm that elevated Gal-3 is associated with cardiovascular disease progression and poor outcome. ...
Role of oxidative stress-related biomarkers in heart failure: galectin 3, α1-antitrypsin and LOX-1: new therapeutic perspective?
0
2020
Systemic inflammation is associated with myocardial fibrosis, diastolic dysfunction, and cardiac hypertrophy in patients with hypertrophic cardiomyopathy
3
2017
... Galectin-3 (Gal-3), a β-galactoside-binding lectin, plays a regulatory role in fibrogenesis. It is widely expressed by various types of inflammatory cells, binds to extracellular glycoproteins, and activates fibroblasts to increase collagen I deposition [95-97]. Growing evidence indicates that high levels of circulating Gal-3 are associated with an increased risk of adverse cardiovascular events, such as HF, myocardial infarction, dilated cardiomyopathy, and fibrogenesis [98-101]. In mice with ischemia/reperfusion (I/R) injury, there was a two-fold increase in plasma Gal-3 levels, and in the ischemic myocardium, Gal-3 was upregulated seven-fold at the mRNA level and 30-fold at the protein level [102]. In addition, that study revealed that plasma Gal-3 concentrations were always higher in DCM/ICM patients with HF than in non-HF subjects, but no trans-cardiac or trans-hepatic concentration gradient of Gal-3 was found. The aforementioned studies were limited in that they did not obtain myocardial biopsies to measure cardiac Gal-3 expression. Moreover, serum Gal-3 is considered to be associated with inflammation and cardiovascular fibrosis. In patients with AMI, serum Gal-3 levels increased immediately after AMI and then declined significantly within 5 days [103]. These data indicate that Gal-3 is closely related to the formation, destabilization, and rupture of plaque. Besides observational studies of Gal-3 levels, several studies have investigated the predictive value of Gal-3. Van Kimmenade et al. observed that an increased circulating Gal-3 level was the best independent predictor of 60-day mortality (odds ratio 10.3, P < 0.01) in patients with acute HF [104]. In patients with coronary artery disease (CAD), Maiolino et al. found that patients with Gal-3 levels in the highest tertile were more prone to death due to cardiovascular causes (25.2%) than those with Gal-3 levels in the intermediate and lower tertiles (13.6% and 7.5%, respectively; P < 0.001), and the plasma Gal-3 cutoff value for cardiovascular death prediction was 27.7 ng/mL [105]. Unfortunately, the loss of 25% of the patients to follow-up may have limited the results of the study. Overall, these findings confirm that elevated Gal-3 is associated with cardiovascular disease progression and poor outcome. ...
... A growing body of evidence has demonstrated that macrophages and monocytes not only play a key role in the occurrence and development of fibrotic responses, but may also mediate the regression of fibrosis [106]. Macrophages and monocytes are capable of producing and excreting a large number of pro-inflammatory mediators, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-4, and IL-6 [107]. Furthermore, a complex regulatory network consisting of these inflammatory factors activates both myofibroblasts and fibrotic pathological processes [107]. This was supported by Kang et al., who treated primary cardiomyocytes with 35 mmol/L glucose for 24 h [108]. The ELISA results showed that the levels of TNF-α, IL-6, and IL-1β and the expression of collagen I and III mRNA were higher than those in the normal control group [108]. These findings suggest that fibrosis is associated with inflammation. In a cross-sectional study of HCM, Fang et al. observed that plasma IL-4, IL-6, and IL-10 levels were positively correlated with diffuse and regional myocardial fibrosis [97]. Unfortunately, owing to the cross-sectional nature of the research, it is impossible to determine whether this association was causal. In addition, in order to study the role of IL-11 signal transduction in cardiovascular fibrosis, Schafer et al. generated IL-11 knockout mice. Compared with wild-type mice, they observed less cardiac fibrosis in knockout mice after either transverse aortic constriction or AngII infusion, suggesting that IL11 is a crucial profibrotic gene [109]. In summary, inflammatory factors are involved in the pathological process of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Galectin-3: a novel mediator of heart failure development and progression
1
2009
... Galectin-3 (Gal-3), a β-galactoside-binding lectin, plays a regulatory role in fibrogenesis. It is widely expressed by various types of inflammatory cells, binds to extracellular glycoproteins, and activates fibroblasts to increase collagen I deposition [95-97]. Growing evidence indicates that high levels of circulating Gal-3 are associated with an increased risk of adverse cardiovascular events, such as HF, myocardial infarction, dilated cardiomyopathy, and fibrogenesis [98-101]. In mice with ischemia/reperfusion (I/R) injury, there was a two-fold increase in plasma Gal-3 levels, and in the ischemic myocardium, Gal-3 was upregulated seven-fold at the mRNA level and 30-fold at the protein level [102]. In addition, that study revealed that plasma Gal-3 concentrations were always higher in DCM/ICM patients with HF than in non-HF subjects, but no trans-cardiac or trans-hepatic concentration gradient of Gal-3 was found. The aforementioned studies were limited in that they did not obtain myocardial biopsies to measure cardiac Gal-3 expression. Moreover, serum Gal-3 is considered to be associated with inflammation and cardiovascular fibrosis. In patients with AMI, serum Gal-3 levels increased immediately after AMI and then declined significantly within 5 days [103]. These data indicate that Gal-3 is closely related to the formation, destabilization, and rupture of plaque. Besides observational studies of Gal-3 levels, several studies have investigated the predictive value of Gal-3. Van Kimmenade et al. observed that an increased circulating Gal-3 level was the best independent predictor of 60-day mortality (odds ratio 10.3, P < 0.01) in patients with acute HF [104]. In patients with coronary artery disease (CAD), Maiolino et al. found that patients with Gal-3 levels in the highest tertile were more prone to death due to cardiovascular causes (25.2%) than those with Gal-3 levels in the intermediate and lower tertiles (13.6% and 7.5%, respectively; P < 0.001), and the plasma Gal-3 cutoff value for cardiovascular death prediction was 27.7 ng/mL [105]. Unfortunately, the loss of 25% of the patients to follow-up may have limited the results of the study. Overall, these findings confirm that elevated Gal-3 is associated with cardiovascular disease progression and poor outcome. ...
Ticagrelor attenuates myocardial ischaemia-reperfusion injury possibly through downregulating galectin-3 expression in the infarct area of rats
0
2018
Galectin-3 and myocardial fibrosis in nonischemic dilated cardiomyopathy
0
2015
Activation of TGF-β1/α-SMA/Col I Profibrotic Pathway in Fibroblasts by Galectin-3 Contributes to Atrial Fibrosis in Experimental Models and Patients
1
2018
... Galectin-3 (Gal-3), a β-galactoside-binding lectin, plays a regulatory role in fibrogenesis. It is widely expressed by various types of inflammatory cells, binds to extracellular glycoproteins, and activates fibroblasts to increase collagen I deposition [95-97]. Growing evidence indicates that high levels of circulating Gal-3 are associated with an increased risk of adverse cardiovascular events, such as HF, myocardial infarction, dilated cardiomyopathy, and fibrogenesis [98-101]. In mice with ischemia/reperfusion (I/R) injury, there was a two-fold increase in plasma Gal-3 levels, and in the ischemic myocardium, Gal-3 was upregulated seven-fold at the mRNA level and 30-fold at the protein level [102]. In addition, that study revealed that plasma Gal-3 concentrations were always higher in DCM/ICM patients with HF than in non-HF subjects, but no trans-cardiac or trans-hepatic concentration gradient of Gal-3 was found. The aforementioned studies were limited in that they did not obtain myocardial biopsies to measure cardiac Gal-3 expression. Moreover, serum Gal-3 is considered to be associated with inflammation and cardiovascular fibrosis. In patients with AMI, serum Gal-3 levels increased immediately after AMI and then declined significantly within 5 days [103]. These data indicate that Gal-3 is closely related to the formation, destabilization, and rupture of plaque. Besides observational studies of Gal-3 levels, several studies have investigated the predictive value of Gal-3. Van Kimmenade et al. observed that an increased circulating Gal-3 level was the best independent predictor of 60-day mortality (odds ratio 10.3, P < 0.01) in patients with acute HF [104]. In patients with coronary artery disease (CAD), Maiolino et al. found that patients with Gal-3 levels in the highest tertile were more prone to death due to cardiovascular causes (25.2%) than those with Gal-3 levels in the intermediate and lower tertiles (13.6% and 7.5%, respectively; P < 0.001), and the plasma Gal-3 cutoff value for cardiovascular death prediction was 27.7 ng/mL [105]. Unfortunately, the loss of 25% of the patients to follow-up may have limited the results of the study. Overall, these findings confirm that elevated Gal-3 is associated with cardiovascular disease progression and poor outcome. ...
Mechanisms responsible for increased circulating levels of galectin-3 in cardiomyopathy and heart failure
2
2018
... Galectin-3 (Gal-3), a β-galactoside-binding lectin, plays a regulatory role in fibrogenesis. It is widely expressed by various types of inflammatory cells, binds to extracellular glycoproteins, and activates fibroblasts to increase collagen I deposition [95-97]. Growing evidence indicates that high levels of circulating Gal-3 are associated with an increased risk of adverse cardiovascular events, such as HF, myocardial infarction, dilated cardiomyopathy, and fibrogenesis [98-101]. In mice with ischemia/reperfusion (I/R) injury, there was a two-fold increase in plasma Gal-3 levels, and in the ischemic myocardium, Gal-3 was upregulated seven-fold at the mRNA level and 30-fold at the protein level [102]. In addition, that study revealed that plasma Gal-3 concentrations were always higher in DCM/ICM patients with HF than in non-HF subjects, but no trans-cardiac or trans-hepatic concentration gradient of Gal-3 was found. The aforementioned studies were limited in that they did not obtain myocardial biopsies to measure cardiac Gal-3 expression. Moreover, serum Gal-3 is considered to be associated with inflammation and cardiovascular fibrosis. In patients with AMI, serum Gal-3 levels increased immediately after AMI and then declined significantly within 5 days [103]. These data indicate that Gal-3 is closely related to the formation, destabilization, and rupture of plaque. Besides observational studies of Gal-3 levels, several studies have investigated the predictive value of Gal-3. Van Kimmenade et al. observed that an increased circulating Gal-3 level was the best independent predictor of 60-day mortality (odds ratio 10.3, P < 0.01) in patients with acute HF [104]. In patients with coronary artery disease (CAD), Maiolino et al. found that patients with Gal-3 levels in the highest tertile were more prone to death due to cardiovascular causes (25.2%) than those with Gal-3 levels in the intermediate and lower tertiles (13.6% and 7.5%, respectively; P < 0.001), and the plasma Gal-3 cutoff value for cardiovascular death prediction was 27.7 ng/mL [105]. Unfortunately, the loss of 25% of the patients to follow-up may have limited the results of the study. Overall, these findings confirm that elevated Gal-3 is associated with cardiovascular disease progression and poor outcome. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Short-term Changes in Gal 3 Circulating Levels After Acute Myocardial Infarction
2
2016
... Galectin-3 (Gal-3), a β-galactoside-binding lectin, plays a regulatory role in fibrogenesis. It is widely expressed by various types of inflammatory cells, binds to extracellular glycoproteins, and activates fibroblasts to increase collagen I deposition [95-97]. Growing evidence indicates that high levels of circulating Gal-3 are associated with an increased risk of adverse cardiovascular events, such as HF, myocardial infarction, dilated cardiomyopathy, and fibrogenesis [98-101]. In mice with ischemia/reperfusion (I/R) injury, there was a two-fold increase in plasma Gal-3 levels, and in the ischemic myocardium, Gal-3 was upregulated seven-fold at the mRNA level and 30-fold at the protein level [102]. In addition, that study revealed that plasma Gal-3 concentrations were always higher in DCM/ICM patients with HF than in non-HF subjects, but no trans-cardiac or trans-hepatic concentration gradient of Gal-3 was found. The aforementioned studies were limited in that they did not obtain myocardial biopsies to measure cardiac Gal-3 expression. Moreover, serum Gal-3 is considered to be associated with inflammation and cardiovascular fibrosis. In patients with AMI, serum Gal-3 levels increased immediately after AMI and then declined significantly within 5 days [103]. These data indicate that Gal-3 is closely related to the formation, destabilization, and rupture of plaque. Besides observational studies of Gal-3 levels, several studies have investigated the predictive value of Gal-3. Van Kimmenade et al. observed that an increased circulating Gal-3 level was the best independent predictor of 60-day mortality (odds ratio 10.3, P < 0.01) in patients with acute HF [104]. In patients with coronary artery disease (CAD), Maiolino et al. found that patients with Gal-3 levels in the highest tertile were more prone to death due to cardiovascular causes (25.2%) than those with Gal-3 levels in the intermediate and lower tertiles (13.6% and 7.5%, respectively; P < 0.001), and the plasma Gal-3 cutoff value for cardiovascular death prediction was 27.7 ng/mL [105]. Unfortunately, the loss of 25% of the patients to follow-up may have limited the results of the study. Overall, these findings confirm that elevated Gal-3 is associated with cardiovascular disease progression and poor outcome. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Utility of amino-terminal pro-brain natriuretic peptide, galectin-3, and apelin for the evaluation of patients with acute heart failure
2
2006
... Galectin-3 (Gal-3), a β-galactoside-binding lectin, plays a regulatory role in fibrogenesis. It is widely expressed by various types of inflammatory cells, binds to extracellular glycoproteins, and activates fibroblasts to increase collagen I deposition [95-97]. Growing evidence indicates that high levels of circulating Gal-3 are associated with an increased risk of adverse cardiovascular events, such as HF, myocardial infarction, dilated cardiomyopathy, and fibrogenesis [98-101]. In mice with ischemia/reperfusion (I/R) injury, there was a two-fold increase in plasma Gal-3 levels, and in the ischemic myocardium, Gal-3 was upregulated seven-fold at the mRNA level and 30-fold at the protein level [102]. In addition, that study revealed that plasma Gal-3 concentrations were always higher in DCM/ICM patients with HF than in non-HF subjects, but no trans-cardiac or trans-hepatic concentration gradient of Gal-3 was found. The aforementioned studies were limited in that they did not obtain myocardial biopsies to measure cardiac Gal-3 expression. Moreover, serum Gal-3 is considered to be associated with inflammation and cardiovascular fibrosis. In patients with AMI, serum Gal-3 levels increased immediately after AMI and then declined significantly within 5 days [103]. These data indicate that Gal-3 is closely related to the formation, destabilization, and rupture of plaque. Besides observational studies of Gal-3 levels, several studies have investigated the predictive value of Gal-3. Van Kimmenade et al. observed that an increased circulating Gal-3 level was the best independent predictor of 60-day mortality (odds ratio 10.3, P < 0.01) in patients with acute HF [104]. In patients with coronary artery disease (CAD), Maiolino et al. found that patients with Gal-3 levels in the highest tertile were more prone to death due to cardiovascular causes (25.2%) than those with Gal-3 levels in the intermediate and lower tertiles (13.6% and 7.5%, respectively; P < 0.001), and the plasma Gal-3 cutoff value for cardiovascular death prediction was 27.7 ng/mL [105]. Unfortunately, the loss of 25% of the patients to follow-up may have limited the results of the study. Overall, these findings confirm that elevated Gal-3 is associated with cardiovascular disease progression and poor outcome. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Galectin-3 predicts long-term cardiovascular death in high-risk patients with coronary artery disease
2
2015
... Galectin-3 (Gal-3), a β-galactoside-binding lectin, plays a regulatory role in fibrogenesis. It is widely expressed by various types of inflammatory cells, binds to extracellular glycoproteins, and activates fibroblasts to increase collagen I deposition [95-97]. Growing evidence indicates that high levels of circulating Gal-3 are associated with an increased risk of adverse cardiovascular events, such as HF, myocardial infarction, dilated cardiomyopathy, and fibrogenesis [98-101]. In mice with ischemia/reperfusion (I/R) injury, there was a two-fold increase in plasma Gal-3 levels, and in the ischemic myocardium, Gal-3 was upregulated seven-fold at the mRNA level and 30-fold at the protein level [102]. In addition, that study revealed that plasma Gal-3 concentrations were always higher in DCM/ICM patients with HF than in non-HF subjects, but no trans-cardiac or trans-hepatic concentration gradient of Gal-3 was found. The aforementioned studies were limited in that they did not obtain myocardial biopsies to measure cardiac Gal-3 expression. Moreover, serum Gal-3 is considered to be associated with inflammation and cardiovascular fibrosis. In patients with AMI, serum Gal-3 levels increased immediately after AMI and then declined significantly within 5 days [103]. These data indicate that Gal-3 is closely related to the formation, destabilization, and rupture of plaque. Besides observational studies of Gal-3 levels, several studies have investigated the predictive value of Gal-3. Van Kimmenade et al. observed that an increased circulating Gal-3 level was the best independent predictor of 60-day mortality (odds ratio 10.3, P < 0.01) in patients with acute HF [104]. In patients with coronary artery disease (CAD), Maiolino et al. found that patients with Gal-3 levels in the highest tertile were more prone to death due to cardiovascular causes (25.2%) than those with Gal-3 levels in the intermediate and lower tertiles (13.6% and 7.5%, respectively; P < 0.001), and the plasma Gal-3 cutoff value for cardiovascular death prediction was 27.7 ng/mL [105]. Unfortunately, the loss of 25% of the patients to follow-up may have limited the results of the study. Overall, these findings confirm that elevated Gal-3 is associated with cardiovascular disease progression and poor outcome. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
Macrophages: master regulators of inflammation and fibrosis
1
2010
... A growing body of evidence has demonstrated that macrophages and monocytes not only play a key role in the occurrence and development of fibrotic responses, but may also mediate the regression of fibrosis [106]. Macrophages and monocytes are capable of producing and excreting a large number of pro-inflammatory mediators, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-4, and IL-6 [107]. Furthermore, a complex regulatory network consisting of these inflammatory factors activates both myofibroblasts and fibrotic pathological processes [107]. This was supported by Kang et al., who treated primary cardiomyocytes with 35 mmol/L glucose for 24 h [108]. The ELISA results showed that the levels of TNF-α, IL-6, and IL-1β and the expression of collagen I and III mRNA were higher than those in the normal control group [108]. These findings suggest that fibrosis is associated with inflammation. In a cross-sectional study of HCM, Fang et al. observed that plasma IL-4, IL-6, and IL-10 levels were positively correlated with diffuse and regional myocardial fibrosis [97]. Unfortunately, owing to the cross-sectional nature of the research, it is impossible to determine whether this association was causal. In addition, in order to study the role of IL-11 signal transduction in cardiovascular fibrosis, Schafer et al. generated IL-11 knockout mice. Compared with wild-type mice, they observed less cardiac fibrosis in knockout mice after either transverse aortic constriction or AngII infusion, suggesting that IL11 is a crucial profibrotic gene [109]. In summary, inflammatory factors are involved in the pathological process of myocardial fibrosis. ...
The pathogenesis of cardiac fibrosis
2
2014
... A growing body of evidence has demonstrated that macrophages and monocytes not only play a key role in the occurrence and development of fibrotic responses, but may also mediate the regression of fibrosis [106]. Macrophages and monocytes are capable of producing and excreting a large number of pro-inflammatory mediators, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-4, and IL-6 [107]. Furthermore, a complex regulatory network consisting of these inflammatory factors activates both myofibroblasts and fibrotic pathological processes [107]. This was supported by Kang et al., who treated primary cardiomyocytes with 35 mmol/L glucose for 24 h [108]. The ELISA results showed that the levels of TNF-α, IL-6, and IL-1β and the expression of collagen I and III mRNA were higher than those in the normal control group [108]. These findings suggest that fibrosis is associated with inflammation. In a cross-sectional study of HCM, Fang et al. observed that plasma IL-4, IL-6, and IL-10 levels were positively correlated with diffuse and regional myocardial fibrosis [97]. Unfortunately, owing to the cross-sectional nature of the research, it is impossible to determine whether this association was causal. In addition, in order to study the role of IL-11 signal transduction in cardiovascular fibrosis, Schafer et al. generated IL-11 knockout mice. Compared with wild-type mice, they observed less cardiac fibrosis in knockout mice after either transverse aortic constriction or AngII infusion, suggesting that IL11 is a crucial profibrotic gene [109]. In summary, inflammatory factors are involved in the pathological process of myocardial fibrosis. ...
... ]. Furthermore, a complex regulatory network consisting of these inflammatory factors activates both myofibroblasts and fibrotic pathological processes [107]. This was supported by Kang et al., who treated primary cardiomyocytes with 35 mmol/L glucose for 24 h [108]. The ELISA results showed that the levels of TNF-α, IL-6, and IL-1β and the expression of collagen I and III mRNA were higher than those in the normal control group [108]. These findings suggest that fibrosis is associated with inflammation. In a cross-sectional study of HCM, Fang et al. observed that plasma IL-4, IL-6, and IL-10 levels were positively correlated with diffuse and regional myocardial fibrosis [97]. Unfortunately, owing to the cross-sectional nature of the research, it is impossible to determine whether this association was causal. In addition, in order to study the role of IL-11 signal transduction in cardiovascular fibrosis, Schafer et al. generated IL-11 knockout mice. Compared with wild-type mice, they observed less cardiac fibrosis in knockout mice after either transverse aortic constriction or AngII infusion, suggesting that IL11 is a crucial profibrotic gene [109]. In summary, inflammatory factors are involved in the pathological process of myocardial fibrosis. ...
Activation of ALDH2 attenuates high glucose induced rat cardiomyocyte fibrosis and necroptosis
3
2020
... A growing body of evidence has demonstrated that macrophages and monocytes not only play a key role in the occurrence and development of fibrotic responses, but may also mediate the regression of fibrosis [106]. Macrophages and monocytes are capable of producing and excreting a large number of pro-inflammatory mediators, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-4, and IL-6 [107]. Furthermore, a complex regulatory network consisting of these inflammatory factors activates both myofibroblasts and fibrotic pathological processes [107]. This was supported by Kang et al., who treated primary cardiomyocytes with 35 mmol/L glucose for 24 h [108]. The ELISA results showed that the levels of TNF-α, IL-6, and IL-1β and the expression of collagen I and III mRNA were higher than those in the normal control group [108]. These findings suggest that fibrosis is associated with inflammation. In a cross-sectional study of HCM, Fang et al. observed that plasma IL-4, IL-6, and IL-10 levels were positively correlated with diffuse and regional myocardial fibrosis [97]. Unfortunately, owing to the cross-sectional nature of the research, it is impossible to determine whether this association was causal. In addition, in order to study the role of IL-11 signal transduction in cardiovascular fibrosis, Schafer et al. generated IL-11 knockout mice. Compared with wild-type mice, they observed less cardiac fibrosis in knockout mice after either transverse aortic constriction or AngII infusion, suggesting that IL11 is a crucial profibrotic gene [109]. In summary, inflammatory factors are involved in the pathological process of myocardial fibrosis. ...
... ]. The ELISA results showed that the levels of TNF-α, IL-6, and IL-1β and the expression of collagen I and III mRNA were higher than those in the normal control group [108]. These findings suggest that fibrosis is associated with inflammation. In a cross-sectional study of HCM, Fang et al. observed that plasma IL-4, IL-6, and IL-10 levels were positively correlated with diffuse and regional myocardial fibrosis [97]. Unfortunately, owing to the cross-sectional nature of the research, it is impossible to determine whether this association was causal. In addition, in order to study the role of IL-11 signal transduction in cardiovascular fibrosis, Schafer et al. generated IL-11 knockout mice. Compared with wild-type mice, they observed less cardiac fibrosis in knockout mice after either transverse aortic constriction or AngII infusion, suggesting that IL11 is a crucial profibrotic gene [109]. In summary, inflammatory factors are involved in the pathological process of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
IL-11 is a crucial determinant of cardiovascular fibrosis
2
2017
... A growing body of evidence has demonstrated that macrophages and monocytes not only play a key role in the occurrence and development of fibrotic responses, but may also mediate the regression of fibrosis [106]. Macrophages and monocytes are capable of producing and excreting a large number of pro-inflammatory mediators, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-4, and IL-6 [107]. Furthermore, a complex regulatory network consisting of these inflammatory factors activates both myofibroblasts and fibrotic pathological processes [107]. This was supported by Kang et al., who treated primary cardiomyocytes with 35 mmol/L glucose for 24 h [108]. The ELISA results showed that the levels of TNF-α, IL-6, and IL-1β and the expression of collagen I and III mRNA were higher than those in the normal control group [108]. These findings suggest that fibrosis is associated with inflammation. In a cross-sectional study of HCM, Fang et al. observed that plasma IL-4, IL-6, and IL-10 levels were positively correlated with diffuse and regional myocardial fibrosis [97]. Unfortunately, owing to the cross-sectional nature of the research, it is impossible to determine whether this association was causal. In addition, in order to study the role of IL-11 signal transduction in cardiovascular fibrosis, Schafer et al. generated IL-11 knockout mice. Compared with wild-type mice, they observed less cardiac fibrosis in knockout mice after either transverse aortic constriction or AngII infusion, suggesting that IL11 is a crucial profibrotic gene [109]. In summary, inflammatory factors are involved in the pathological process of myocardial fibrosis. ...
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
T-cell-dependent fibrosis in the mdx dystrophic mouse
1
2000
... MMPs and miRNAs also play a regulatory role in the process of inflammatory fibrosis. Tissue injury induces activation of inflammatory mediators and recruitment of inflammatory cells. Inflammatory cells promote fibrosis by producing cytokines/chemokines that regulate MMPs [110]. MMP-9, the most widely documented protease in the inflammatory process, is involved in the initial stage of myocardial injury. The increase in MMP-9 levels is related to the invasion of necrotic tissue by inflammatory cells, particularly polymorphonuclear neutrophils and activated satellite cells [111]. Moreover, in the cardiovascular system, miRNAs control the expression of some inflammatory factors. For example, miR-155 controls the expression of SHIP1 and SOCS1, the key regulators of the inflammatory response in macrophages [112]. Elevated miR-21 expression in macrophages inhibited the production of TNF-α and the up-regulation of IL-10, respectively [113]. Overall, these inflammatory markers are associated with fibrosis. ...
Activation of hypoxia-inducible factor 1 in skeletal muscle cells after exposure to damaged muscle cell debris
1
2011
... MMPs and miRNAs also play a regulatory role in the process of inflammatory fibrosis. Tissue injury induces activation of inflammatory mediators and recruitment of inflammatory cells. Inflammatory cells promote fibrosis by producing cytokines/chemokines that regulate MMPs [110]. MMP-9, the most widely documented protease in the inflammatory process, is involved in the initial stage of myocardial injury. The increase in MMP-9 levels is related to the invasion of necrotic tissue by inflammatory cells, particularly polymorphonuclear neutrophils and activated satellite cells [111]. Moreover, in the cardiovascular system, miRNAs control the expression of some inflammatory factors. For example, miR-155 controls the expression of SHIP1 and SOCS1, the key regulators of the inflammatory response in macrophages [112]. Elevated miR-21 expression in macrophages inhibited the production of TNF-α and the up-regulation of IL-10, respectively [113]. Overall, these inflammatory markers are associated with fibrosis. ...
MicroRNA-155 is induced during the macrophage inflammatory response
1
2007
... MMPs and miRNAs also play a regulatory role in the process of inflammatory fibrosis. Tissue injury induces activation of inflammatory mediators and recruitment of inflammatory cells. Inflammatory cells promote fibrosis by producing cytokines/chemokines that regulate MMPs [110]. MMP-9, the most widely documented protease in the inflammatory process, is involved in the initial stage of myocardial injury. The increase in MMP-9 levels is related to the invasion of necrotic tissue by inflammatory cells, particularly polymorphonuclear neutrophils and activated satellite cells [111]. Moreover, in the cardiovascular system, miRNAs control the expression of some inflammatory factors. For example, miR-155 controls the expression of SHIP1 and SOCS1, the key regulators of the inflammatory response in macrophages [112]. Elevated miR-21 expression in macrophages inhibited the production of TNF-α and the up-regulation of IL-10, respectively [113]. Overall, these inflammatory markers are associated with fibrosis. ...
Engulfment of apoptotic cells by macrophages: a role of microRNA-21 in the resolution of wound inflammation
1
2014
... MMPs and miRNAs also play a regulatory role in the process of inflammatory fibrosis. Tissue injury induces activation of inflammatory mediators and recruitment of inflammatory cells. Inflammatory cells promote fibrosis by producing cytokines/chemokines that regulate MMPs [110]. MMP-9, the most widely documented protease in the inflammatory process, is involved in the initial stage of myocardial injury. The increase in MMP-9 levels is related to the invasion of necrotic tissue by inflammatory cells, particularly polymorphonuclear neutrophils and activated satellite cells [111]. Moreover, in the cardiovascular system, miRNAs control the expression of some inflammatory factors. For example, miR-155 controls the expression of SHIP1 and SOCS1, the key regulators of the inflammatory response in macrophages [112]. Elevated miR-21 expression in macrophages inhibited the production of TNF-α and the up-regulation of IL-10, respectively [113]. Overall, these inflammatory markers are associated with fibrosis. ...
Activation of orphan receptors by the hormone relaxin
1
2002
... The development of myocardial fibrosis also involves factors such as relaxin-1 (RLN1), growth hormone secretagogue receptor (GSHR), ADAMTS-1, and transient receptor potential melastatin 7 (TRPM7). RLN, a polypeptide hormone, is secreted by cardiac cells [114]. In a cross-sectional study, the average circulating RLN1 levels in the HFrEF patient population were markedly higher than those in healthy control subjects (702 ± 283 pg/mL vs. 44 ± 27 pg/mL), with elevated RLN1 levels accompanied by a decrease in heart fibrosis [115]. This finding promotes the study of RLN1 as a potential anti-fibrosis target. GSHR is an orexigenic hormone with newly defined cardiovascular effects. In a mouse model of isoproterenol-induced myocardial fibrosis, GHSR deficiency exacerbated the expression of myofibroblast trans-differentiation marker genes, suggesting that GHSR may be a surrogate indicator of the need for intervention in myocardial fibrosis [116]. ADAMTS-1, a metalloprotease with proteolytic activity, also has the ability to cleave the N-terminal propeptide of collagen. In mice with viral heart disease (VHD), Li et al. observed that the collagen volume fraction increased significantly, accompanied by elevated expression of ADAMTS-1 mRNA in cardiac tissue (P < 0.001) [117]. Moreover, TRPM7 is capable of promoting the proliferation and differentiation of fibroblasts and increasing the synthesis of extracellular matrix proteins [118]. ...
Circulating Relaxin-1 Level Is a Surrogate Marker of Myocardial Fibrosis in HFrEF
2
2019
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
... The development of myocardial fibrosis also involves factors such as relaxin-1 (RLN1), growth hormone secretagogue receptor (GSHR), ADAMTS-1, and transient receptor potential melastatin 7 (TRPM7). RLN, a polypeptide hormone, is secreted by cardiac cells [114]. In a cross-sectional study, the average circulating RLN1 levels in the HFrEF patient population were markedly higher than those in healthy control subjects (702 ± 283 pg/mL vs. 44 ± 27 pg/mL), with elevated RLN1 levels accompanied by a decrease in heart fibrosis [115]. This finding promotes the study of RLN1 as a potential anti-fibrosis target. GSHR is an orexigenic hormone with newly defined cardiovascular effects. In a mouse model of isoproterenol-induced myocardial fibrosis, GHSR deficiency exacerbated the expression of myofibroblast trans-differentiation marker genes, suggesting that GHSR may be a surrogate indicator of the need for intervention in myocardial fibrosis [116]. ADAMTS-1, a metalloprotease with proteolytic activity, also has the ability to cleave the N-terminal propeptide of collagen. In mice with viral heart disease (VHD), Li et al. observed that the collagen volume fraction increased significantly, accompanied by elevated expression of ADAMTS-1 mRNA in cardiac tissue (P < 0.001) [117]. Moreover, TRPM7 is capable of promoting the proliferation and differentiation of fibroblasts and increasing the synthesis of extracellular matrix proteins [118]. ...
GHSR Deficiency Exacerbates Cardiac Fibrosis: Role in Macrophage Inflammasome Activation and Myofibroblast Differentiation
2
2019
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
... The development of myocardial fibrosis also involves factors such as relaxin-1 (RLN1), growth hormone secretagogue receptor (GSHR), ADAMTS-1, and transient receptor potential melastatin 7 (TRPM7). RLN, a polypeptide hormone, is secreted by cardiac cells [114]. In a cross-sectional study, the average circulating RLN1 levels in the HFrEF patient population were markedly higher than those in healthy control subjects (702 ± 283 pg/mL vs. 44 ± 27 pg/mL), with elevated RLN1 levels accompanied by a decrease in heart fibrosis [115]. This finding promotes the study of RLN1 as a potential anti-fibrosis target. GSHR is an orexigenic hormone with newly defined cardiovascular effects. In a mouse model of isoproterenol-induced myocardial fibrosis, GHSR deficiency exacerbated the expression of myofibroblast trans-differentiation marker genes, suggesting that GHSR may be a surrogate indicator of the need for intervention in myocardial fibrosis [116]. ADAMTS-1, a metalloprotease with proteolytic activity, also has the ability to cleave the N-terminal propeptide of collagen. In mice with viral heart disease (VHD), Li et al. observed that the collagen volume fraction increased significantly, accompanied by elevated expression of ADAMTS-1 mRNA in cardiac tissue (P < 0.001) [117]. Moreover, TRPM7 is capable of promoting the proliferation and differentiation of fibroblasts and increasing the synthesis of extracellular matrix proteins [118]. ...
Expression of ADAMTS-1 mRNA in myocardium of viral heart disease mice and its clinical significance
2
2019
... Biomarkers of myocardial fibrosis (collagen synthesis, breakdown, metabolism).
Biomarkers
Studied Condition
Test Location
Main findings
Reference
PICP
HCM patients
Plasma
PICP ↑
[10]
Hypertension patients
Serum
PICP ↑
[11]
HF rats
Plasma
PICP ↓
[12]
HF patients
Serum
PICP ↑
[13]
PINP
HFrEF patients
Serum
PINP ↑
[18]
MI rats
Plasma
PINP ↑
[19]
HCM patients
Serum
Not predictive in fibrosis
[20]
PIIINP
DCM patients
Serum
PIIINP ↑
[25]
HFrEF patients
Serum
PIIINP ↑, positively associated with diastolic function
[26]
CoA patients
Serum
PIIINP ↑, positively associated with LVMI and RWT
[27]
Hypertension patients
Serum
PIIINP ↑, inversely associated with diastolic functions
[28]
MI rats
Serum
PIIINP ↑
[29]
CITP
HF patients
Serum
CITP ↑
[20]
HF and AF patients
Serum
CITP ↑
[32-33]
DCM patients
Serum
Not predictive in fibrosis
[34]
AMI patients
Serum
CITP ↑, predicts cardiovascular mortality
[35]
MMPs
HCM patients
Serum
MMP-2 ↓, MMP-9 was positively associated with fibrosis
[44]
systolic HF patients
Serum
MMP-1↑
[45-46]
DM rats
Myocardial tissues
MMP2↓, MMP7, MMP11, MMP13, MMP16↑
[47]
TIMPs
2K1C hypertension rats
Myocardial tissues
TIMP-4↑
[58]
TTC patients
Serum
TIMP-1 and MMP-2 ↑
[59]
HF patients
Serum
TIMP-1↑
[60]
TGF-β
MI mice
Myocardial tissues
anti-TGF-β treated: collagen production ↓, matrix-metalloproteinase ↑
anti-TGF-βtreated: perivascular and interstitial fibrosis ↓
[68]
Smads
TAC mice
Cardiac tissues
Smad3 or Smad2/3 deletion: cardiac fibrosis↓
[72]
Smad3-deficient mice
Myocardial tissues
Collagen content and deposition ↓
[73]
CTGF
DCM rats
Myocardial tissues
CTGF/CCN2 gene silencing: cardiac function ↑, myocardial fibrosis and left ventricular hypertrophy ↓
[81]
MI rats
Myocardial tissues
CTGF ↑
[83]
Chronic HF patients
Plasma
CTGF ↑
[84]
Corin
HF patients
Plasma
Corin ↓
[85]
HF canines
Myocardial tissues
Corin ↓
[89]
HCM disease
Cardiomyocytes
Corin↑
[90]
EndoMT
ISO-induced fibrosis
Myocardial tissues
α-SMA and vimentin: ↑ with a peak on day 3, and then gradually↓
[92]
DCM rats
Myocardial tissues
a-SMA, fibronectin and vimentin↑
[93]
In vitro
Mouse embryonic fibroblasts
Vimentin deletion: WF-A did not change half-lives of collagen mRNAs
[94]
Gal-3
Myocardial I/R injury rats
Myocardial tissues
Gal-3 ↑
[102]
AMI patients
Serum
Gal-3 ↑ after AMI and then ↓ within 5 days
[103]
Acute HF patients
Serum
Gal-3 ↑
[104]
CAD patients
Plasma
Gal-3 is positively correlated with cardiovascular deaths
[105]
TNF-α and interleukin
Cardiomyocyte fibrosis
Cardiomyocytes
TNF-α, IL-6 IL-1β levels and collagen I and III mRNA expressions ↑
[108]
HCM patients
Plasma
IL-4, IL-6, and IL-10 ↑
[97]
IL-11 knockout mice
Myocardial tissues
Fibrosis↓ after either transverse aortic constriction or AngII infusion
[109]
Other Molecules
HFrEF patients
Serum
RLN1↑
[115]
ISO-induced cardiac fibrosis
Myocardial tissues
GSHR deletion: myocardial fibrosis ↑
[116]
VHD mice
Myocardial tissues
ADAMTS-1↑
[117]
Abbreviations: PICP, C-terminal Propeptide of Procollagen Type I; PINP, Procollagen Type I N-terminal Propeptide; PIIINP, Procollagen Type III Amino-terminal Propeptide; CITP, C-terminal Telopeptide of Collagen Type I; MMPs, Matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinase; TGF-β, Transforming growth factor-β; CTGF, connective tissue growth factor; EndoMT, endothelial to mesenchymal transition; Gal-3, Galectin-3 protein; TNF-α, tumor necrosis factor -α; HCM, hypertrophic cardiomyopathy; HF, heart failure; HFrEF, heart failure and reduced ejection fraction; MI, myocardial infarction; DCM, dilated cardiomyopathy; CoA, coarctation of the aorta; AF, atrial fibrillation; AMI, acute myocardial infarction; DM, diabetic mice; 2K1C:two-kidney one-clip; TTC, takotsubo cardiomyopathy; TAC, transverse aortic constriction; I/R, ischemia/reperfusion; ISO, isoproterenol; CAD, coronary artery disease; VHD, viral heart disease; LVMI, left ventricular mass index; RWT, relative wall thickness; RLN1, relaxin-1; GSHR, growth hormone secretagogue receptor ...
... The development of myocardial fibrosis also involves factors such as relaxin-1 (RLN1), growth hormone secretagogue receptor (GSHR), ADAMTS-1, and transient receptor potential melastatin 7 (TRPM7). RLN, a polypeptide hormone, is secreted by cardiac cells [114]. In a cross-sectional study, the average circulating RLN1 levels in the HFrEF patient population were markedly higher than those in healthy control subjects (702 ± 283 pg/mL vs. 44 ± 27 pg/mL), with elevated RLN1 levels accompanied by a decrease in heart fibrosis [115]. This finding promotes the study of RLN1 as a potential anti-fibrosis target. GSHR is an orexigenic hormone with newly defined cardiovascular effects. In a mouse model of isoproterenol-induced myocardial fibrosis, GHSR deficiency exacerbated the expression of myofibroblast trans-differentiation marker genes, suggesting that GHSR may be a surrogate indicator of the need for intervention in myocardial fibrosis [116]. ADAMTS-1, a metalloprotease with proteolytic activity, also has the ability to cleave the N-terminal propeptide of collagen. In mice with viral heart disease (VHD), Li et al. observed that the collagen volume fraction increased significantly, accompanied by elevated expression of ADAMTS-1 mRNA in cardiac tissue (P < 0.001) [117]. Moreover, TRPM7 is capable of promoting the proliferation and differentiation of fibroblasts and increasing the synthesis of extracellular matrix proteins [118]. ...
Effects of angiotensin II on transient receptor potential melastatin 7 channel function in cardiac fibroblasts
1
2015
... The development of myocardial fibrosis also involves factors such as relaxin-1 (RLN1), growth hormone secretagogue receptor (GSHR), ADAMTS-1, and transient receptor potential melastatin 7 (TRPM7). RLN, a polypeptide hormone, is secreted by cardiac cells [114]. In a cross-sectional study, the average circulating RLN1 levels in the HFrEF patient population were markedly higher than those in healthy control subjects (702 ± 283 pg/mL vs. 44 ± 27 pg/mL), with elevated RLN1 levels accompanied by a decrease in heart fibrosis [115]. This finding promotes the study of RLN1 as a potential anti-fibrosis target. GSHR is an orexigenic hormone with newly defined cardiovascular effects. In a mouse model of isoproterenol-induced myocardial fibrosis, GHSR deficiency exacerbated the expression of myofibroblast trans-differentiation marker genes, suggesting that GHSR may be a surrogate indicator of the need for intervention in myocardial fibrosis [116]. ADAMTS-1, a metalloprotease with proteolytic activity, also has the ability to cleave the N-terminal propeptide of collagen. In mice with viral heart disease (VHD), Li et al. observed that the collagen volume fraction increased significantly, accompanied by elevated expression of ADAMTS-1 mRNA in cardiac tissue (P < 0.001) [117]. Moreover, TRPM7 is capable of promoting the proliferation and differentiation of fibroblasts and increasing the synthesis of extracellular matrix proteins [118]. ...
MicroRNAs in the onset and development of cardiovascular disease
1
2014
... MicroRNAs (miRNAs) are small, noncoding RNAs that regulate gene expression after transcription, and play a critical role in heart function and pathology [119]. The miRNA family regulates gene expression by inducing target mRNA destabilization or inhibiting protein translation [120]. The marked cardiospecific capacity of some miRNAs is beneficial for ameliorating tissue remodeling. ...
Circulating microRNAs as biomarkers for diffuse myocardial fibrosis in patients with hypertrophic cardiomyopathy
1
2015
... MicroRNAs (miRNAs) are small, noncoding RNAs that regulate gene expression after transcription, and play a critical role in heart function and pathology [119]. The miRNA family regulates gene expression by inducing target mRNA destabilization or inhibiting protein translation [120]. The marked cardiospecific capacity of some miRNAs is beneficial for ameliorating tissue remodeling. ...
Myocardial and circulating levels of microRNA-21 reflect left ventricular fibrosis in aortic stenosis patients
2
2013
... The relationship between miR-21 and myocardial fibrosis has been extensively studied. miRNAs target not only single genes, but also overall networks that contribute to biological function. For example, in patients with aortic stenosis (AS), myocardial and plasmatic miR-21 levels predicted collagen type I, collagen type III, and fibronectin expression by targeting RECK, PDCD4, and TGF-β-signaling factors [121]. In animal models of myocardial infarction, miR-21 promoted the transformation of cardiac fibroblasts (CFs) to myofibroblasts, and increased myocardial fibrosis in vivo by targeting Jagged1 [122]. In addition, the overexpression of miR-21 in allogeneic mice activated the fibrosis gene program and promoted the differentiation of monocytes to fibroblasts via the phosphatase and tensin homologue/activator protein 1 regulatory axis (PTEN/AP-1) [123]. Intriguingly, Szemraj-Rogucka et al., in a study of 13 left ventricular non-compaction patients (LVNC), found that plasma levels of miR-21, miR-29a, miR-30d, and miR-133a were both significantly elevated, suggesting that all four miRNAs may serve as biomarkers of myocardial fibrosis [124]. Unfortunately, this study was performed using a small sample size, which may have biased the results. ...
... Biomarkers of myocardial fibrosis (gene transcription).
miR-21 promotes cardiac fibroblast-to-myofibroblast transformation and myocardial fibrosis by targeting Jagged1
2
2018
... The relationship between miR-21 and myocardial fibrosis has been extensively studied. miRNAs target not only single genes, but also overall networks that contribute to biological function. For example, in patients with aortic stenosis (AS), myocardial and plasmatic miR-21 levels predicted collagen type I, collagen type III, and fibronectin expression by targeting RECK, PDCD4, and TGF-β-signaling factors [121]. In animal models of myocardial infarction, miR-21 promoted the transformation of cardiac fibroblasts (CFs) to myofibroblasts, and increased myocardial fibrosis in vivo by targeting Jagged1 [122]. In addition, the overexpression of miR-21 in allogeneic mice activated the fibrosis gene program and promoted the differentiation of monocytes to fibroblasts via the phosphatase and tensin homologue/activator protein 1 regulatory axis (PTEN/AP-1) [123]. Intriguingly, Szemraj-Rogucka et al., in a study of 13 left ventricular non-compaction patients (LVNC), found that plasma levels of miR-21, miR-29a, miR-30d, and miR-133a were both significantly elevated, suggesting that all four miRNAs may serve as biomarkers of myocardial fibrosis [124]. Unfortunately, this study was performed using a small sample size, which may have biased the results. ...
... Biomarkers of myocardial fibrosis (gene transcription).
... The relationship between miR-21 and myocardial fibrosis has been extensively studied. miRNAs target not only single genes, but also overall networks that contribute to biological function. For example, in patients with aortic stenosis (AS), myocardial and plasmatic miR-21 levels predicted collagen type I, collagen type III, and fibronectin expression by targeting RECK, PDCD4, and TGF-β-signaling factors [121]. In animal models of myocardial infarction, miR-21 promoted the transformation of cardiac fibroblasts (CFs) to myofibroblasts, and increased myocardial fibrosis in vivo by targeting Jagged1 [122]. In addition, the overexpression of miR-21 in allogeneic mice activated the fibrosis gene program and promoted the differentiation of monocytes to fibroblasts via the phosphatase and tensin homologue/activator protein 1 regulatory axis (PTEN/AP-1) [123]. Intriguingly, Szemraj-Rogucka et al., in a study of 13 left ventricular non-compaction patients (LVNC), found that plasma levels of miR-21, miR-29a, miR-30d, and miR-133a were both significantly elevated, suggesting that all four miRNAs may serve as biomarkers of myocardial fibrosis [124]. Unfortunately, this study was performed using a small sample size, which may have biased the results. ...
... Biomarkers of myocardial fibrosis (gene transcription).
Circulating microRNAs as biomarkers for myocardial fibrosis in patients with left ventricular non-compaction cardiomyopathy
2
2019
... The relationship between miR-21 and myocardial fibrosis has been extensively studied. miRNAs target not only single genes, but also overall networks that contribute to biological function. For example, in patients with aortic stenosis (AS), myocardial and plasmatic miR-21 levels predicted collagen type I, collagen type III, and fibronectin expression by targeting RECK, PDCD4, and TGF-β-signaling factors [121]. In animal models of myocardial infarction, miR-21 promoted the transformation of cardiac fibroblasts (CFs) to myofibroblasts, and increased myocardial fibrosis in vivo by targeting Jagged1 [122]. In addition, the overexpression of miR-21 in allogeneic mice activated the fibrosis gene program and promoted the differentiation of monocytes to fibroblasts via the phosphatase and tensin homologue/activator protein 1 regulatory axis (PTEN/AP-1) [123]. Intriguingly, Szemraj-Rogucka et al., in a study of 13 left ventricular non-compaction patients (LVNC), found that plasma levels of miR-21, miR-29a, miR-30d, and miR-133a were both significantly elevated, suggesting that all four miRNAs may serve as biomarkers of myocardial fibrosis [124]. Unfortunately, this study was performed using a small sample size, which may have biased the results. ...
... Biomarkers of myocardial fibrosis (gene transcription).
... Another well-researched miRNA is miR-29, which demonstrates reduced expression under cardiac stress conditions, and thus may promote more extracellular matrix protein production by “derepression” of elastin, genes encoding collagens, and fibrillin [125]. Heid et al. found that overexpression of the miR-29 family counteracted the physiological accumulation of oxidative damage during aging, thereby protecting the heart against the detrimental effects of fibrosis [126]. In patients with pathological hypertrophy, TaqMan quantitative polymerase chain reaction showed a marked decrease in cardiac miR-29a and miR-29c, suggesting that miR-29 expression has been linked to extracellular matrix remodeling in cardiac hypertrophy [127]. MiR-29 is also a good example of a potential therapeutic target. For example, infusion of anti-miR-29 into cardiac hypertrophy model mice could prevent the expression of fibrosis markers, such as Col1a1, Col1a2, and Col3a1 [128]. Together, the miR-29 family plays a key role in pathological hypertrophy of the myocardium and fibrosis. ...
Age-dependent increase of oxidative stress regulates microRNA-29 family preserving cardiac health
1
2017
... Another well-researched miRNA is miR-29, which demonstrates reduced expression under cardiac stress conditions, and thus may promote more extracellular matrix protein production by “derepression” of elastin, genes encoding collagens, and fibrillin [125]. Heid et al. found that overexpression of the miR-29 family counteracted the physiological accumulation of oxidative damage during aging, thereby protecting the heart against the detrimental effects of fibrosis [126]. In patients with pathological hypertrophy, TaqMan quantitative polymerase chain reaction showed a marked decrease in cardiac miR-29a and miR-29c, suggesting that miR-29 expression has been linked to extracellular matrix remodeling in cardiac hypertrophy [127]. MiR-29 is also a good example of a potential therapeutic target. For example, infusion of anti-miR-29 into cardiac hypertrophy model mice could prevent the expression of fibrosis markers, such as Col1a1, Col1a2, and Col3a1 [128]. Together, the miR-29 family plays a key role in pathological hypertrophy of the myocardium and fibrosis. ...
Extracellular matrix secretion by cardiac fibroblasts: role of microRNA-29b and microRNA-30c
2
2013
... Biomarkers of myocardial fibrosis (gene transcription).
... Another well-researched miRNA is miR-29, which demonstrates reduced expression under cardiac stress conditions, and thus may promote more extracellular matrix protein production by “derepression” of elastin, genes encoding collagens, and fibrillin [125]. Heid et al. found that overexpression of the miR-29 family counteracted the physiological accumulation of oxidative damage during aging, thereby protecting the heart against the detrimental effects of fibrosis [126]. In patients with pathological hypertrophy, TaqMan quantitative polymerase chain reaction showed a marked decrease in cardiac miR-29a and miR-29c, suggesting that miR-29 expression has been linked to extracellular matrix remodeling in cardiac hypertrophy [127]. MiR-29 is also a good example of a potential therapeutic target. For example, infusion of anti-miR-29 into cardiac hypertrophy model mice could prevent the expression of fibrosis markers, such as Col1a1, Col1a2, and Col3a1 [128]. Together, the miR-29 family plays a key role in pathological hypertrophy of the myocardium and fibrosis. ...
Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling
2
2017
... Biomarkers of myocardial fibrosis (gene transcription).
... Another well-researched miRNA is miR-29, which demonstrates reduced expression under cardiac stress conditions, and thus may promote more extracellular matrix protein production by “derepression” of elastin, genes encoding collagens, and fibrillin [125]. Heid et al. found that overexpression of the miR-29 family counteracted the physiological accumulation of oxidative damage during aging, thereby protecting the heart against the detrimental effects of fibrosis [126]. In patients with pathological hypertrophy, TaqMan quantitative polymerase chain reaction showed a marked decrease in cardiac miR-29a and miR-29c, suggesting that miR-29 expression has been linked to extracellular matrix remodeling in cardiac hypertrophy [127]. MiR-29 is also a good example of a potential therapeutic target. For example, infusion of anti-miR-29 into cardiac hypertrophy model mice could prevent the expression of fibrosis markers, such as Col1a1, Col1a2, and Col3a1 [128]. Together, the miR-29 family plays a key role in pathological hypertrophy of the myocardium and fibrosis. ...
Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling
2
2013
... Biomarkers of myocardial fibrosis (gene transcription).
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress
2
2018
... Biomarkers of myocardial fibrosis (gene transcription).
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
Up-regulation of microRNA-203 inhibits myocardial fibrosis and oxidative stress in mice with diabetic cardiomyopathy through the inhibition of PI3K/Akt signaling pathway via PIK3CA
2
2019
... Biomarkers of myocardial fibrosis (gene transcription).
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
Astragaloside IV inhibits cardiac fibrosis via miR-135a-TRPM7-TGF-β/Smads pathway
3
2020
... Biomarkers of myocardial fibrosis (gene transcription).
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
microRNA-122 down-regulation may play a role in severe myocardial fibrosis in human aortic stenosis through TGF-β1 up-regulation
0
2014
MiR-195 inhibits myocardial fibrosis in hypertensive rats by regulating TGFβ1-Smad3 signaling pathway
Overexpression of microRNA-202-3p protects against myocardial ischemia-reperfusion injury through activation of TGF-β1/Smads signaling pathway by targeting TRPM6
2
2019
... Biomarkers of myocardial fibrosis (gene transcription).
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
Relations between circulating microRNAs (miR-21, miR-26, miR-29, miR-30 and miR-133a), extracellular matrix fibrosis and serum markers of fibrosis in dilated cardiomyopathy
1
2017
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
Association of miR-197-5p, a Circulating Biomarker for Heart Failure, with Myocardial Fibrosis and Adverse Cardiovascular Events among Patients with Stage C or D Heart Failure
2
2018
... Biomarkers of myocardial fibrosis (gene transcription).
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction)
2
2016
... Biomarkers of myocardial fibrosis (gene transcription).
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-β1 pathway
2
2012
... Biomarkers of myocardial fibrosis (gene transcription).
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
MicroRNA-150 Inhibits the Activation of Cardiac Fibroblasts by Regulating c-Myb
2
2016
... Biomarkers of myocardial fibrosis (gene transcription).
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
Loss of miR-144 signaling interrupts extracellular matrix remodeling after myocardial infarction leading to worsened cardiac function
2
2018
... Biomarkers of myocardial fibrosis (gene transcription).
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
Effects of miR-29a and miR-101a Expression on Myocardial Interstitial Collagen Generation After Aerobic Exercise in Myocardial-infarcted Rats
2
2017
... Biomarkers of myocardial fibrosis (gene transcription).
... Other cardiomyocyte-associated miRNAs, such as miR-26, miR-30, miR-135, and miR-208, are also involved in cardiac fibrosis. miR-1 expression reverses pressure-induced myocardial hypertrophy and prevents cardiac remodeling by secretion of the protein fibullin-2 (FBLN2), which is related to extracellular matrix remodeling [129]. miR-378 is secreted from cardiomyocytes at the early stage of cardiac remodeling after mechanical stress and inhibits excessive cardiac fibrosis through paracrine mechanisms [130]. Overexpression of miR-203 prevented cardiac collagen type I and type III expression in DCM mice by targeting PIK3CA through inactivation of the PI3K/Akt signaling pathway [131]. As upstream molecules of TGF-β, miR-135a, miR-122, miR-195, miR-202-3p, and miR-328 also participate in the process of fibrosis by regulating the expression of myocardial collagen [132-136]. Rubiś et al. found that baseline serum levels of miR-21, miR-26, miR-29, and miR-30 were significantly different in DCM patients than in controls, and that MMP-2 levels were strongly associated with all of the microRNAs studied [137]. In a prospective study, elevated baseline plasma miR-197-5P levels were considered to be associated with myocardial fibrosis in patients with end-stage HF, and during the average 937-day follow-up period, 22 patients (27.5%) had major adverse cardiac events, including 19 deaths and three cardiac transplantations [138]. Additionally, multiple studies have found evidence implicating cardiac interstitial fibrosis in the deterioration of cardiac function in myocardial infarction. In AMI, miR-208 and miR-499 mediate cardioblast?cardiomyocyte transformation and fast/slow muscle fiber specification at the late cardiogenic stages [139]. In addition, 4 weeks after coronary artery ligation in rats, miR-101 and miR-150 were decreased in the peri-infarct area and were expressed in cardiac fibroblasts [140, 141]. Moreover, miR-144 knockout mice demonstrated impaired late remodeling after MI, which was reflected by elevated total cardiac collagen content [142]. Interestingly, intermittent aerobic exercise could enhance the expression of cardiac miR-101a in rats with MI, and miR-101a was associated with decreased expression of fibrotic genes, such as Tgfb, fos, Smad2/3, Col1A1, and Col3A1 [143]. These findings indicate that cardiac miRNAs (miR-208, miR-101, miR-150, and miR-144) play a central role in fibrosis after MI. Overall, miRNAs may become a potential therapeutic target for myocardial fibrosis (miRNAs are briefly summarized in Table 2). ...
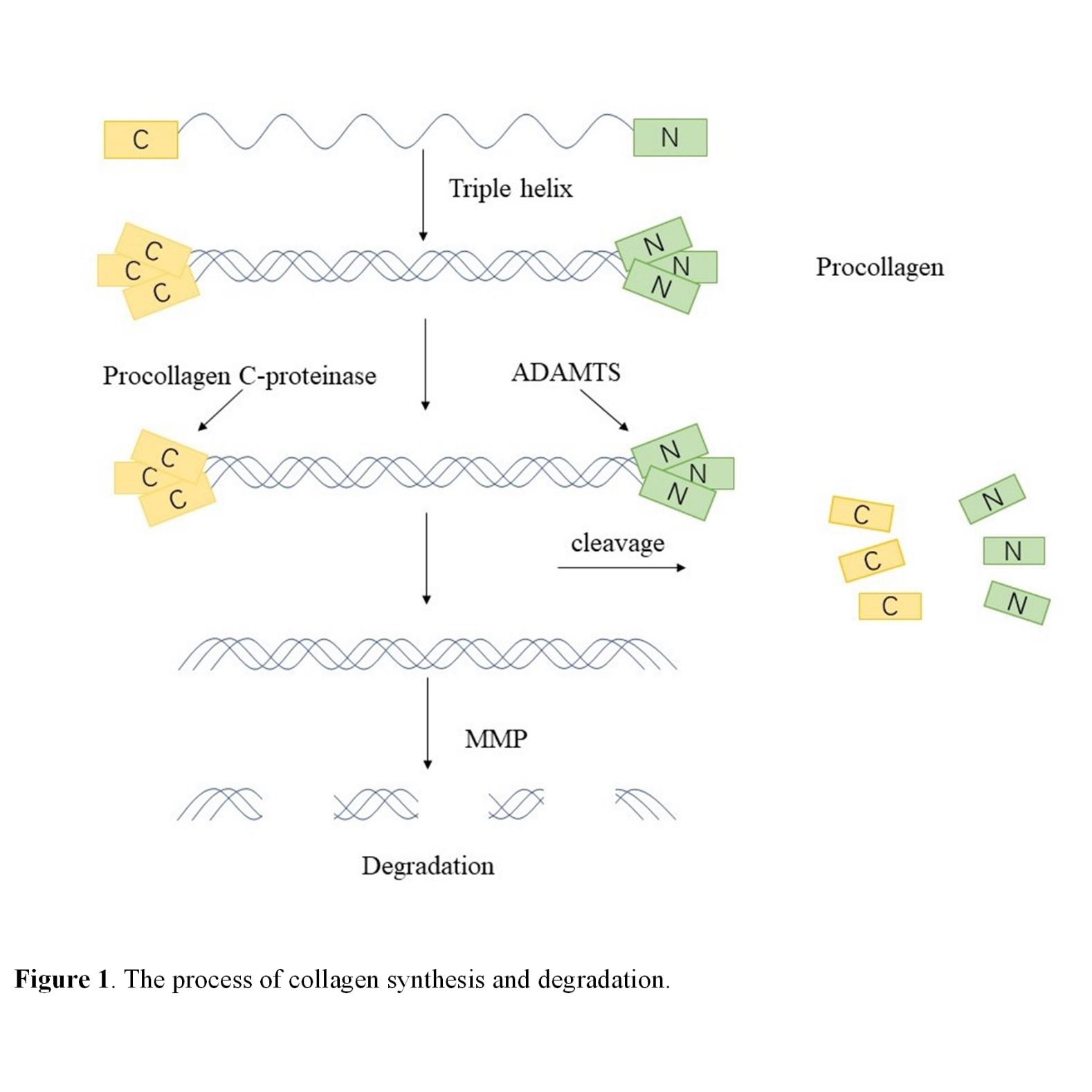
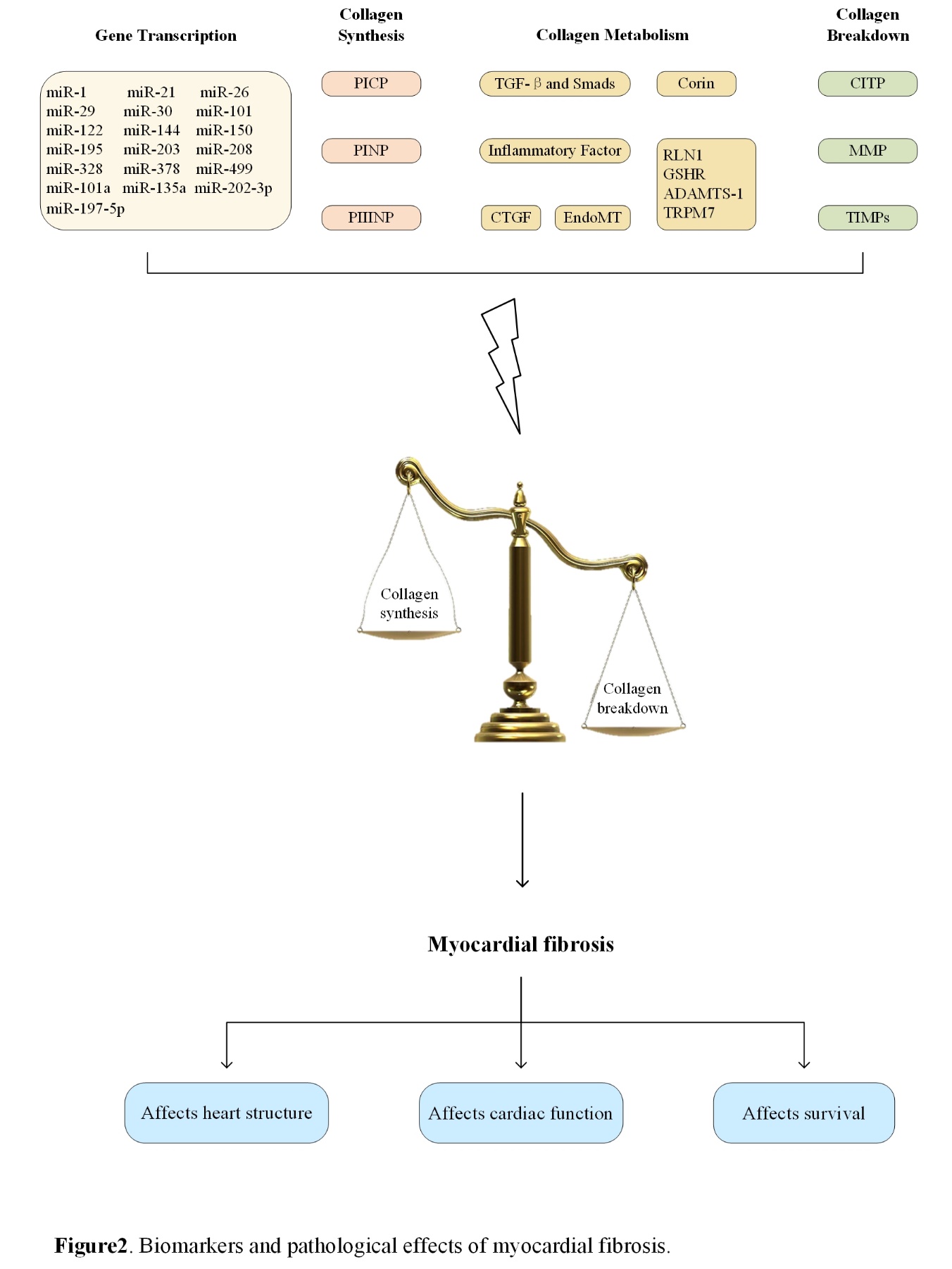
{kind=link}
{kind=link}
{kind=link}
{kind=link}



