Yang Yibo, Li Tianyi, Li Zhibo, Liu Ning, Yan Youyou, Liu Bin*
通讯作者: Correspondence should be addressed to: Dr. Bin Liu, Department of Cardiology, The Second Hospital of Jilin University, 218 Ziqiang Road, Changchun 130041, China. Email: liubin3333@vip.sina.com.Correspondence should be addressed to: Dr. Bin Liu, Department of Cardiology, The Second Hospital of Jilin University, 218 Ziqiang Road, Changchun 130041, China. Email: liubin3333@vip.sina.com.
收稿日期:2019-03-27
修回日期: 2019-05-16
接受日期: 2019-05-18
网络出版日期: 2020-03-09
版权声明:
2020 This is an Open Access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
展开
Abstract
Cardiovascular disease is the leading cause of mortality worldwide, and mitochondrial dysfunction is the primary contributor to these disorders. Recent studies have elaborated on selective autophagy-mitophagy, which eliminates damaged and dysfunctional mitochondria, stabilizes mitochondrial structure and function, and maintains cell survival and growth. Numerous recent studies have reported that mitophagy plays an important role in the pathogenesis of various cardiovascular diseases. This review summarizes the mechanisms underlying mitophagy and advancements in studies on the role of mitophagy in cardiovascular disease.
YangYibo, LiTianyi, LiZhibo, LiuNing, YanYouyou, LiuBin. Role of Mitophagy in Cardiovascular Disease[J]. Aging and Disease, 2020, 11(2): 419-437 https://doi.org/10.14336/AD.2019.0518
1. Introduction
Owing to the epidemiological shift in the 20th century, infectious disease-related death and disability have decreased; however, they have increased rapidly owing to noninfectious diseases including cardiovascular disease (CVD), which have some of the highest mortality and morbidity rates. After the 1930s, mortalities due to CVD exceeded those due to tumors, tuberculosis, and pneumonia. CVD became a leading cause of mortality in developed countries [1]. CVD includes heart and vascular diseases and is a major public health concern and cause of mortality [2]. The prevalence of CVD has increased notwithstanding improvements in treatment and management methods. Age-standardized mortality of CVD has decreased by 14.5% over the past two decades [3]. Nonetheless, with an increase in the incidence of cardiovascular disease and changes in the disease spectrum, the incidence of CVD and CVD-associated mortality are expected to increase worldwide in future decades. Therefore, prevention and treatment of cardiovascular disease is extremely urgent [4]. In CVD, the final and the most prominent phenomenon is cell death caused by harmful stimuli and aging. Mitochondria provide energy for cellular metabolism and are involved in cell death. They participate in cell proliferation, apoptosis, signal transduction, and calcium homeostasis. Hence, the mitochondrial steady state is essential for cellular function [5-7].
Mitochondria are the most abundant organelles in cardiomyocytes and are very important for the maintenance of normal cell function. Mitophagy is selective autophagy wherein cells eliminate damaged or dysfunctional mitochondria, which is essential for the regulation of mitochondrial homeostasis [8-10]. Mitophagy was first proposed by Lemasters et al. in 2005. Since then, numerous diseases have been associated with mitophagy [11]. Recent studies revealed that mitophagy contributes to the pathophysiology of CVD. Mitophagy regulates cardiovascular activity through several pathways [12-22]. The present review discusses mitophagy and its underlying mechanisms, methods to assess mitophagy and its involvements in CVD.
Figure 1. The mechanisms underlying mitophagy. (A) An overview of the mechanisms underlying mitophagy. (B) Step1: Phagophores are formed by the isolated membrane and LC3. Step2: Thereafter, through LC3 adaptors and LC3 receptors, damaged mitochondria can be recognized and form mitophagosomes. The detailed mechanism can be divided into six stages. a. CHDH accumulates on the outer mitochondrial membrane (OMM) and interacts with p62 and binds with LC3. b. PINK1 accumulates on the OMM, phosphorylates Parkin and Mfn2, thus recruiting Parkin to the OMM, and Parkin helps generate ubiquitin chains on the OMM, which can recognize p62 and bind with LC3. c. PINK1 phosphorylates ubiquitin on the OMM and LC3 adapters can bind with it. d. LC3 directly recognizes BNIP3 or Nix through LIR, and phosphorylation of LIR in BNIP3 promotes the interaction between BNIP3 and LC3. e. Dephosphorylation of FUNDC1 restores its ability to interact with LC3 through LIR. f. AMBRA1, Bcl2L13, and cardiophospholipids directly recognize LC3 through LIR. Step3: Mitophagosomes and lysosomes fuse into mitolysosomes.
2. Mitophagy
Mitochondria regulate calcium steady state, signal transmission between organelles, and oxidative phosphorylation for ATP biosynthesis. Mitochondrial DNA (mtDNA) is present in the matrix or membrane and does not combine with histones. Therefore, mtDNAs are relatively vulnerable to reactive oxygen species (ROS) and influenced by mutations. Mitochondria are more susceptible to oxidative damage than other organelles [23-25]. Dysfunctional and damaged mitochondria generate excess ROS, which damage proteins, membrane lipids, nucleic acids, adjacent mitochondria and release proapoptotic proteins into the cytosol, eventually causing cell death [25-28]. Hence, prompt and appropriate elimination of damaged or aging mitochondria is vital for normal cell growth. This process is executed by mitophagy [11].
Autophagy involves the degradation of the protoplasm by lysosomes, resulting in macrophage-, microphage-, and chaperone-mediated autophagy. Selective autophagy occurs in response to nutrient deficiency, whereas nonspecific autophagy is caused by cell aging or a response to damaged cell components. Mitophagy is a selective autophagy pathway involving the elimination of abnormal mitochondria and maintains mitochondrial homeostasis [16, 29-32].
2.1 Mitophagy and related factors
Mitophagy is divided into three steps: formation of phagocytic vesicles, recognition of damaged mitochondrial components and compartmentation into mitochondrial phagocytic vesicles, and fusion of mitochondrial phagocytic vesicles and lysosomes into mitolysosomes [16, 33, 34](Fig. 1). The mitophagy pathway was first identified in yeast in 2004. Mitochondrial outer membrane protein autophagy-related gene 32 (Atg32) recruits core autophagy molecules to the mitochondria for selective clearance [35-37]; however, no homologs have been reported in the human genome thus far. The subsequent section summarizes the mechanisms underlying mitophagy in accordance with the three aforementioned stages.
Step 1. Formation of phagocytic vesicles (Phagophores)
Phagocytic vesicles are isolated membranous structures in the endoplasmic reticulum (ER) of mammalian cells. The formation of phagocytic vesicles is regulated by an evolutionarily conserved protein (Atg) system [35, 37-39]. Atg6 recruits other Atg proteins to the isolation membrane, thus warranting the Atg5-Atg12 and LC3-phosphatidylethanolamine conjugation systems. Covalent conjugation between Atg12 and Atg5 resembles ubiquitination, wherein Atg12 is initially activated by Atg7, and then transferred to Atg10. Atg12 is then covalently conjugated with Atg5 at a lysine residue and Atg10 is released. Finally, the Atg12-Atg5 complex interacts non-covalently with Atg16, which then initiates the elongation of the membrane by recruiting LC3-phosphatidylethanolamine [39]. LC3 is a surface molecule on phagocytic vesicles. Phagocytic vesicles are formed by combining LC3-I with phosphatidylethanolamine to produce lipid LC3-II [33, 40, 41].
Step 2. Recognition of damaged mitochondrial components and compartmentation into mitochondrial phagocytic vesicles (Mitophagosomes)
Mitophagy is a type of selective organellar autophagy in mammalian cells. Identification of phagocytic vesicles and their combination with damaged mitochondria have received increasing attention in studies on mitophagy [33, 42]. Damaged mitochondria are recognized by LC3 adapters or LC3 receptors. First, we address the recognition of impaired mitochondria by LC3 adapters.
PINK1 and Parkin
In mammalian cells, PINK1/Parkin-mediated mitophagy is the most extensively characterized mitophagic phenomenon thus far [43]. PINK1 is a serine/threonine kinase with an N-terminal mitochondrial targeting signal [44]. In intact mitochondria, PINK1 is recruited to the mitochondria by translational enzymes on the outer mitochondrial membrane (OMM) and the endometrial complex, anchored within the mitochondrial endometrium, and continuously degraded by protease and peptidase. During mitochondrial depolarization, however, translocation of PINK1 across to the inner mitochondrial membrane (IMM) is inhibited. PINK1 is not degraded but aggregates at the outer membrane [16, 42].
PINK1 utilizes its own ubiquitin-like domain to function like ubiquitin. Activated PINK1 phosphorylates Parkin (a cytoplasmic E3 ubiquitin ligase) at T175, thus activating Parkin, which in turn aggregates in the OMM. However, activated PINK1 also phosphorylates Mfn2, which can then serve as a Parkin receptor in cardiomyocyte mitochondria. This process also induces Parkin aggregation on the OMM [43, 45-47]. Phospho-parkin helps generate ubiquitin chains on the OMM proteins through polyubiquitination, thus amplifying the signal, and these ubiquitin chains serve as LC3 adapter recognition sites, which bind to LC3 and initiate mitophagy [48, 49].
PINK1 may also be recruited in a Parkin-independent manner. PINK1 phosphorylates the mitochondrial ubiquitin on OMM at S65. LC3 adapters such as optineurin and NDP52 recognize the damaged mitochondria and ubiquitin phosphorylated at S65 through the ubiquitin-binding region [48].
Choline dehydrogenase (CHDH)
CHDH oxidizes choline to betaine aldehyde in the mitochondria. In normal mitochondria, CHDH is unanchored at both the OMM and IMM. In depolarized mitochondria, CHDH accumulates on the OMM and its FAD/NAD-binding domain 1 interacts with the Phox and Bem1 domains in cytoplasmic p62/SQSTM1, thus recruiting p62/SQSTM1 at damaged mitochondria and binding with LC3 [42, 50].
In summary, the impaired mitochondrial recognition process mediated by the LC3 adapter is divided into the ubiquitin-associated type (PINK1/Parkin) and the ubiquitin-independent type (CHDH). Five LC3 adapters have been identified including OPTN, p62/SQSTM1, NBR1, NDP52, and TAX1BP, which mediate the recognition and phagocytosis of damaged mitochondria by joining LC3-coated phagocytic vesicles to label damaged mitochondria [33, 42]. Further, we discuss the LC3 reporter-mediated recognition of impaired mitochondria.
Nix and BNIP3
Nix and BNIP3 are OMM proteins that interact with LC3 or its homologs through the LC3-interacting region (LIR) to form mitophagy receptors. BNIP3 and Nix are hypoxia-induced tail-anchored proteins that bind to the OMM via the carboxyl transmembrane domain. The N-terminal extends into the cytoplasm and is linked to LC3-related molecules[51-53]. BNIP3-dependent mitophagy requires drp1 to induce mitochondrial division. Inhibition of this process or drp1 knockout blocks hypoxia-induced mitophagy. Nix protein levels increase after erythrocyte maturation. Nix knockout inhibits the loss of the mitochondrial membrane potential and the isolation of mitochondrial autophagosomes. Nix-dependent mitophagy is induced by the loss of membrane potential caused by carbonyl cyanide m-chlorophenylhydrazone treatment. Therefore, Nix-mediated loss of membrane proteins is important in mitophagy [54].
FUNDC1
FUNDC1 is an OMM protein, which integrates with LC3 under hypoxia via its LIR structure. During starvation, mitophagy does not depend on FUNDC1; therefore, it specifically induces hypoxia [55]. FUNDC1 is expressed under normoxic conditions; however, FUNDC1 and LC3 interact under hypoxic conditions upon dephosphorylation at Y18 [56]. Furthermore, FUNDC1 is a mitochondrial receptor for unc-51-like autophagy activating kinase 1 (ULK1), which in turn is translocated to depolarized mitochondria and FUNDC1 phosphorylated at S17 and promotes mitophagy [56]. Phosphoglycerate mutase family member 5 phosphatase is located at the mitochondria and dephosphorylates FUNDC1 at S13 under hypoxic conditions or upon treatment with carbonyl cyanide p-trifluoromethoxy phenylhydrazone. The latter promotes the interaction of the enzyme with LC3 and induces mitophagy [57].
LC3 receptors BNIP3/Nix and FUNDC1 have been extensively characterized in cardiovascular studies. BCL2-like13 (Bcl2L13), autophagy/Beclin 1 regulator 1 (AMBRA1), and cardio phospholipids serve as LC3 receptors during mitophagy [16, 42, 58]. They localize at the OMM thus eliminating the need for mitophagic receptors to mark the damaged mitochondria. Rather, they directly interact with LC3 via the LIR and identify and engulf the damaged mitochondria [42, 48, 50].
Step 3. Mitochondrial phagocytic vesicles and lysosomes fuse into mitolysosomes
Phagocytic vesicles fuse with damaged mitochondria to form mitolysosomes. Thereafter, various hydrolytic enzymes in the lysosome digest the damaged mitochondrial components, thereby executing the final step of mitophagy. Recent studies on mitophagy primarily focused on the formation, recognition, and interaction of phagocytic vesicles with damaged mitochondria. The detailed mechanism underlying the binding of phagocytic vesicles with lysosomes shall not be described in this review.
2.2 Research methods
Discussed herein are several widely used techniques to assess mitophagy.
Transmission electron microscopy (TEM)
The most prominent phenomenon in autophagy is the formation of a double-layered membrane around the cytosol and/or organelles. This structure fuses with the lysosome, its contents are released, and a single-layer membrane is formed. Therefore, early and late autophagy are easily distinguished by observing double- and single-membrane structures. Consistent with cell autophagy, early mitophagy potentially results in packaging of the double-membrane structure, which can be readily identified on the basis of its unique structure-mitochondrial crest. During late autophagy, mitochondria are transported to the lysosomes and can be identified from their density or residual structure. TEM is currently the best method to detect mitophagy. It is the "gold standard" method to directly detect autophagy [59, 60]. However, TEM has several limitations. Electron microscopy requires extensive sample preparation. Cell damage potentially resulting from human error interferes with observations. Moreover, mitochondrial structures are difficult to discern during later stages of autophagy. Therefore, this method has low sensitivity.
Western blotting
A commonly used method to detect autophagy proteins is western blotting. Autophagy can be detected on the basis of the ratio of the LC3B II/I type marker protein and the extent of P62 degradation [61]. Mitophagy is accompanied by the degradation of several mitochondrial proteins; hence, it may also be evaluated by measuring their levels. Mitochondrial degradation occurs through the autophagy pathway, and cytosolic LC3 protein is converted from type I to type II via conjugation with phosphatidylethanolamine. Therefore, LC3 detection is also a potential method of detecting autophagy. The use of autophagy inhibitors including Bafilomycin A1 (Baf A1) and hydroxychloroquine (HCQ) is also a prominent method of detecting autophagy [41, 62]. Recent studies have reported that the OMM, IMM, and matrix proteins are degraded via mitophagy, whereas certain proteins regulating mitochondrial division and migration are degraded by proteasomes or ATP-dependent protease pathways. TIM23, TOM20, Hsp60, and electron transport chain (ETC)-related proteins are commonly used as mitophagy markers [63]. P62 can be recruited individually, since it is highly ubiquitinated after mitochondrial damage. P62 is involved in numerous signal transduction pathways and its function is complex. Therefore, the role of P62 in mitophagy detection is controversial [64, 65].
Fluorescence labeling
GFP-LC3 is often used as an autophagosome marker. Following plasmid transfection, autophagosome formation may be assessed using a fluorescence microscope. Similarly, plasmids containing fluorescent markers or specific antibodies against certain mitochondrial proteins including TOM20 and cytochrome C can be used to label mitochondria. Several commercialized mitochondrial dyes such as MitoTracker and TMRE (Tetramethylrhodamine ethyl ester) are also potentially applicable. In general, mitochondria and autophagosomes/lysosomes can be labeled separately and the colocalization of mitochondria, autophagosomes, and lysosomes may be detected via confocal laser microscopy to detect mitophagy. LC3 fused with fluorescent proteins, such as GFP and RFP, helps observe mitochondrial incorporation into autophagosomes, accompanied with the extension of the isolation membrane through time-lapse microscopic imaging of live cells. GFP fluorescence is attenuated under acidic conditions; hence, GFP-LC3 does not label autolysosomes. However, RFP is resistant to the acidic environment of lysosomes. By harnessing the distinct properties of GFP and RFP, GFP-RFP-tandem tagging LC3 is a potentially useful tool to quantify mitophagy and helps distinguish autophagosomes and autolysosomes. However, this evaluation may overlook the previously reported mitophagy mediated via an LC3-independent mechanism [66-68]. Recently, a mitochondrial probe MitoTimer was developed, which is a red fluorescent protein Ds Red "mutant" and encodes a redox sensitive mitochondrial targeted protein that switches from green to red fluorescence upon oxidation [69]. Therefore, MitoTimer is an ROS-dependent fluorophore. Furthermore, changes in the transition from green to red fluorescence are affected by temperature, oxygen, and light exposure; hence, this assay must be performed combinatorially with other methods to accurately determine the causes of changes in red or green fluorescence levels. Moreover, MitoTimer is a very useful tool to indirectly quantify mitophagy [70]. This assay quantifies several pathophysiological parameters of mitochondria, accurately assesses their health status, and plays potentially important roles in future studies [62, 71, 72]. Moreover, mitoKeima is also a fluorophore that can help quantify mitophagy. Changes in the emission wavelength of mitoKeima depend on the acidity (red color) or neutrality (green color) of the pH of the microenvironment, e.g., during the transfer from an autophagosome (green color) to a lysosome (red color). Hence, mitoKeima is a powerful pH-dependent fluorophore that is more specific for monitoring mitophagy than MitoTimer [73, 74].
Determination of mitochondrial mass
Mitophagy is accompanied by a reduction in mitochondrial mass; therefore, direct detection of changes in mitochondrial mass helps detect mitophagy. Mitochondria are labeled with mitochondrial dyes or corresponding antibodies, and the number of mitochondria per cell is determined via flow cytometry. RT-PCR may be used to quantify mtDNA [75-77]. These methods directly quantify mitochondrial mass. Simultaneously, mitochondrial mass is regulated via numerous regulatory mechanisms, and mitochondrial biogenesis and degradation are reportedly two of the most prominent regulatory mechanisms. Therefore, the decline in mitochondrial mass may not only be affected by mitophagy but also a process involving multiple regulatory mechanisms. Hence, determination of mitochondrial mass is an adjunct method of detecting mitophagy [78].
2.3 Factors inducing mitophagy
During cell growth and development, numerous endogenous or exogenous stimulants can alter the degree of mitophagy. Therefore, mitophagy is involved in various pathophysiological processes.
Mitophagy and stress
In the cardiovascular system, mitophagy exerts protective effects in response to cellular starvation. Mitophagy is activated during starvation to regulate mitochondrial quality and quantity through elimination of damaged or redundant mitochondria. Mitophagy is enhanced during cellular hypoxia. Acetylcholine reduces the damage caused by hypoxia/reoxygenation by increasing mitophagy in H9C2 cells. Violent exercise alters mitophagy levels by adaptively regulating mitochondrial function [79-81].
Mitophagy and the cellular defense
During intracellular pathogenic infections, mitophagy is induced to defend against pathogens. Pathogens also induce mitophagy and disrupt mitochondrial homeostasis. For example, sepsis induces reversible cardiac mitochondrial injury, and recovery is reportedly associated with mitophagy based on morphological and biochemical evidence, suggesting that mitophagy is activated and plays an important role during cardiac recovery from sepsis [82, 83].
Mitophagy and the maintenance of cellular homeostasis
In normal cells, mitophagy may have a housekeeping function and renew mitochondrial components. Mitophagy participates in metabolic remodeling and is closely associated with inflammatory response factors. For example, orally administered polyaminospermine in mice enhances mitophagy, inhibits subclinical inflammation, and increases longevity. Changes in the degree of mitophagy modulate stem cell activity and chronic cardiomyocyte inflammation [19, 84].
Mitophagy and the regulation of cell growth and development
Mitophagy alters the oxidative respiratory chain activity by regulating mitochondrial quantity and quality, thus contributing to the regulation of cell growth and development. For example, macrophage fibrosis factor (Mff)-deficient mice died of dilated cardiomyopathy at 13 weeks. Mitophagy was significantly elevated in their mutated tissues [84, 85].
Mitophagy and aging
Cellular aging has a prominent mitochondrial basis. Mitophagy reportedly mediates various degenerative diseases and contributes to their pathophysiology and treatment. For example, mitophagy is disrupted in the heart of aged individuals and in senescent cells and DOX-treated hearts and cells [19, 86].
3. Mitophagy and cardiovascular disease
Since mitophagy is involved in numerous prominent pathophysiological phenomena, it contributes to CVD pathogenesis.
3.1 Ischemic heart disease
Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99].
3.2 Diabetic cardiomyopathy
Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117].
3.3 Heart failure
Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure
3.4 Hypertension
Hypertension is a systemic disease with a high incidence and is closely associated with cardiovascular, cerebrovascular, kidney, and other diseases [130]. There is no direct evidence for the role of mitophagy in the pathophysiology of hypertension. Complications of hypertension such as atherosclerosis, myocardial I/R injury and hypertrophy, and heart failure are closely associated with cardiomyocyte and endothelial mitochondrial dysfunction [131]. Insufficient energy supply, kinetic imbalance, oxidative damage, abnormal signal transduction, and mitochondrial dysfunction caused by genetic mutations are risk factors for hypertension [132-134]. Myocardial hypertrophy associated with hypertension reportedly altered cardiomyocyte phenotype, function, and energy metabolism. These changes were reflected in the utilization of metabolic substrates and the function of the ETC, ATP synthesis, and other processes [135]. ROS regulate vascular structure and tension remodeling. An increase or retention of ROS may cause vasoconstriction, decrease vascular diastolic function, damage smooth muscle cells, promote vascular inflammation and remodeling, increase peripheral vascular resistance, and aggravate hypertension [136]. Mfn1, Mfn2, and OPA1 (optic atrophy 1) mRNA levels, which are associated with mitochondrial dynamics, were decreased in hypertensive rat heart models. Hence, mitochondrial fragmentation is increased in hypertension [137]. Mitochondrial division is a compensatory mechanism maintaining cardiac contraction in hypertension and exacerbating ventricular wall thickening [138]. Hypertension is suggested to alter mitochondrial function, energy metabolism, and mitochondrial dynamics in cardiomyocytes [131]. Clearance of mitochondrial dysfunction is closely associated with mitophagy. Therefore, elucidation of the role of mitophagy in the occurrence and development of hypertension may be important to investigate the pathophysiology of this condition and novel treatments for this condition.
3.5 Atherosclerosis
Atherosclerosis (AS) is a chronic disease characterized by lipid accumulation, vascular smooth muscle cell proliferation, apoptosis, and local inflammation [139]. The pathogenesis of atherosclerosis is complex and includes abnormal lipid metabolism, inflammation, endothelial injury, oxidative stress, and other mechanisms [140, 141]. In the pathophysiology of atherosclerosis, plaque tissue stability determines disease occurrence and progression [142]. In plaque macrophages, vascular smooth muscle cells, and endothelial cells, mitophagy clears the damaged mitochondria, stabilizes the mitochondrial function in the damaged cells, reduces cell damage, maintains plaque integrity, and prevents disease progression caused by plaque tissue rupture [143]. Oxidized low-density lipoprotein (ox-LDL) has been widely used to generate atherosclerosis models because it is an atherosclerotic agent. In vascular smooth muscle cells, ox-LDL disrupts mitochondrial structure, increases ROS levels, and induces mitochondrial dysfunction [144]. Furthermore, Ox-LDL activates mitophagy, which eliminates nonfunctional and damaged mitochondria. After the addition of mitophagy inhibitors, mitophagy and its related pathway proteins are significantly decreased, and cell apoptosis is significantly increased [145]. Mitophagy protects against atherosclerotic stress-induced apoptosis in human vascular smooth muscle cells (VSMCs) [146-148]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques.
3.6 Arrhythmia
Arrhythmia is a prominent aspect of CVD. It may occur individually or in association with other cardiovascular disease. In a healthy heart, coordinated electrical propagation supports cardiac function; upon failure of such a cardiac action potential, cardiac arrhythmia occurs [149]. Although there is no direct evidence of the role of mitophagy in arrhythmia, extensive evidence indicates that mitochondria play a functional role in arrhythmia. Mitochondria are involved in arrhythmia owing to their ability to produce ATP and ROS [150]. Mitochondrial dysfunction reduces ATP production, affects cardiac electrical conduction, thus altering sarcolemmal K+ fluxes via ATP-sensitive potassium channels [151]. However, excessive mitochondrial ROS production can introduce heterogeneity in cardiac action potentials [152]. Alterations in ATP and ROS levels are based on a vicious cycle: during mitochondrial dysfunction, the mitochondrial membrane potential reduced, thus decreasing ATP and increasing ROS levels; thereafter, further mitochondrial dysfunction is induced, with the mitochondrial membrane potential reduced, with a concomitant reduction in ATP generation. This vicious cycle causes electrophysiological alterations, thereby causing arrhythmia [150, 153]. Proper mitophagy can eliminate damaged mitochondria, maintain mitochondrial homeostasis in cells, and prevent damage to hazardous substances, thus further disrupting cellular phenomena. During arrhythmia, changes in ATP and ROS levels caused by mitochondrial damage play an important role in mitophagy. Therefore, we can boldly speculate that mitophagy eliminates damaged mitochondria and regulates ROS and ATP levels to a certain extent, thereby inhibiting or decelerating the occurrence of arrhythmia.
3.7 Stroke
Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163].
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1).
4.1 MicroRNAs
MicroRNAs are small, single-stranded, noncoding RNA molecules which inhibit the translation of target mRNAs or induce their degradation [164]. MiR-410 is reportedly significantly upregulated in an I/R injury model of human adult cardiomyocytes (HACMs), which presented decreased mitochondrial function and loss of mitophagy. MiR-410 overexpression reportedly decreased cell viability, ATP generation, mitochondrial membrane potential, and mitophagy. In contrast, miR-410 downregulation had the opposite effect. MiR-410 directly targeted HSPB1 and regulated its activity and mitophagy [165]. MiR-137 is reportedly significantly upregulated under hypoxia, wherein it downregulates Nix and FUNDC1 and impairs mitophagy [166]. In lipid toxicity and diabetic mouse models, miR-133a was reportedly downregulated and Nix was upregulated. MiR-133a upregulation inhibited Nix translation, regulated mitochondrial function, and stabilized mitochondrial membrane potential [167].
4.2 Clinical drugs and chemical reagents
Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180].
4.3 Signaling pathways
Several signaling pathways regulate molecules that control mitophagy. In a mouse atherosclerosis model established with ox-LDL, NR4A1 was significantly upregulated and controlled Parkin activation through posttranscriptional CaMKII modification. Thus, it activated Parkin-mediated mitophagy. Excessive mitophagy reduced mitochondrial quality which, in turn, caused energy shortage and mitochondrial dysfunction [145]. Furthermore, blockade of the MAPK-ERK-CREB signaling pathway upregulated NR4A1 and repressed Mfn2-mediated mitophagy, thus expanding the area of cerebral infarction and increasing neuronal apoptosis [181]. In cardiomyocytes, damaged mitochondria are sequestered in rab5-positive early endosomes via the ESCRT mechanism, which depends on Parkin, and the loss of rab5 function increases the susceptibility of embryonic fibroblasts and cardiomyocytes to cell death [182]. In a mouse ischemic model, activation of the p53-TIGAR axis downregulated BNIP3, inhibited mitophagy, caused the accumulation of damaged mitochondria, and weakened the cardioprotective effect [183]. The JNK signaling pathway is a key regulator of the FOXO3a transcription factor, which promotes BNIP3 expression in heart failure models. JNK influences the degree of mitophagy by regulating the level of BNIP3[184].
4.4 Activators/inhibitors and gene knock in/out
Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96].
4.5 Environmental stimuli
Certain environmental stimuli can inhibit or promote mitophagy and alter the content of the proteins related to this phenomenon. Mild hypothermia downregulated Parkin in hippocampal neurons in a cardiac arrest model, diminishing mitophagy in the neurons, protecting mitochondria, and improving neurological function after cardiac arrest [194]. Hypoxic preconditioning induced FUNDC1-dependent activation of mitophagy and decreased I/R-induced cardiac injury [96]. Myocardial mitochondrial function adapts to stress during acute exercise and manifests as significant upregulation of the mitophagy-related protein BNIP3, which stimulates mitophagy and minimizes myocardial injury [80]. Simultaneously, exercise preconditioning significantly suppresses exhaustive exercise-induced hypoxia-ischemia injuries through upregulated parkin-dependent mitophagy [195]. Acidic postconditioning-induced mitophagy renders the brain resistant to ischemic stroke through recruitment of PARK2 to mitochondria [196]. Remote ischemic post-conditioning promoted mitophagy via Parkin upregulation and inhibited oxidative stress responses, thus mitigating cerebral I/R injury in a rat model of stroke [197].
Figure 2. The graphical abstract. Mitophagy plays an important role in cardiovascular disease, and the degree of mitophagy can be detected via TEM, western blotting, fluorescence labeling, and mitochondrial mass determination, and the related molecular mechanism depends on PINK1/Parkin, CHDH, Nix/BNIP3, FUNDC1, etc. Mitophagy is related with certain physiological and pathological phenomena including stress, cellular defense, maintenance of cellular homeostasis, regulation of cell growth and development, and aging; these phenomena are also involved in the pathogenesis of cardiovascular diseases including ischemic heart disease, diabetic cardiomyopathy, heart failure, hypertension, atherosclerosis, arrhythmia, and stroke, and these diseases are closely associated with mitophagy. Therefore, certain factors including microRNAs, clinical drugs and chemical reagents, signaling pathways, activators/inhibitors and gene knock in/out, and environmental stimuli can regulate the level of mitophagy to alter the progression of these diseases.
5. Conclusion and Outlook
Mitochondrial injury and dysfunction are prominent pathophysiological mechanisms of CVD. Mitophagy potentially mitigates or alleviates these disorders and maintains cellular stability. The underlying mechanisms are complex, interrelated, and regulated by numerous stimulating factors. Several factors induce mitophagy during cellular growth, development, aging, and death. Alterations in the degree of mitophagy may alter the course of cardiovascular disease. Therefore, mitophagy is a potential therapeutic target for cardiovascular disease (Fig. 2). Nevertheless, it is yet unclear whether mitophagy retards or accelerates damage in CVD pathogenesis. Furthermore, it is unclear whether mitophagy is mediated by the unique functions of different molecules. Further studies are required to investigate whether the mechanisms underlying mitophagy induced by various molecules have mutual effects and the role of mitophagy in CVD, thus leading to advancements in the development of clinical diagnoses and treatments.
Acknowledgments
This study was supported by grants from the National R&D Program of China (No.2016YFC1301100), National Natural Science Foundation of China (No. 81570250, NO. 31600728), and Science and Technology Department of Jilin Province (No.201603059YY).
Conflicts of Interest
The authors declare that they have no conflict of interests.
DehghanM, MenteA, ZhangX, SwaminathanS, LiW, MohanV, et al. (2017).
Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): a prospective cohort study
NaghaviM, AbajobirA, AbbafatiC, AbbasK, Abd-AllahF, AberaS, et al. (2017).
Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the Global Burden of Disease Study 2016
Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy
FerreeAW, TrudeauK, ZikE, BenadorIY, TwigG, GottliebRA, et al. (2013).
MitoTimer probe reveals the impact of autophagy, fusion, and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age
To Explore the Protective Mechanism of PTEN-Induced Kinase 1 (PINK1)/Parkin Mitophagy-Mediated Extract of Periplaneta Americana on Lipopolysaccharide-Induced Cardiomyocyte Injury
FengY, MadungweNB, da Cruz JunhoCV, BopassaJC (2017).
Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy
DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways
GakidouE, AfshinA, AbajobirA, AbateK, AbbafatiC, AbbasK, et al. (2018).
Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017
YuP, ZhangJ, YuS, LuoZ, HuaF, YuanL, et al. (2015).
Protective Effect of Sevoflurane Postconditioning against Cardiac Ischemia/Reperfusion Injury via Ameliorating Mitochondrial Impairment, Oxidative Stress and Rescuing Autophagic Clearance
ZhangX, YuanY, JiangL, ZhangJ, GaoJ, ShenZ, et al. (2014).
Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy
RenJ, YangL, ZhuL, XuX, CeylanAF, GuoW, et al. (2017).
Akt2 ablation prolongs life span and improves myocardial contractile function with adaptive cardiac remodeling: role of Sirt1-mediated autophagy regulation
Reducing the Global Burden of Cardiovascular Disease, Part 1: The Epidemiology and Risk Factors
1
2017
... Owing to the epidemiological shift in the 20th century, infectious disease-related death and disability have decreased; however, they have increased rapidly owing to noninfectious diseases including cardiovascular disease (CVD), which have some of the highest mortality and morbidity rates. After the 1930s, mortalities due to CVD exceeded those due to tumors, tuberculosis, and pneumonia. CVD became a leading cause of mortality in developed countries [1]. CVD includes heart and vascular diseases and is a major public health concern and cause of mortality [2]. The prevalence of CVD has increased notwithstanding improvements in treatment and management methods. Age-standardized mortality of CVD has decreased by 14.5% over the past two decades [3]. Nonetheless, with an increase in the incidence of cardiovascular disease and changes in the disease spectrum, the incidence of CVD and CVD-associated mortality are expected to increase worldwide in future decades. Therefore, prevention and treatment of cardiovascular disease is extremely urgent [4]. In CVD, the final and the most prominent phenomenon is cell death caused by harmful stimuli and aging. Mitochondria provide energy for cellular metabolism and are involved in cell death. They participate in cell proliferation, apoptosis, signal transduction, and calcium homeostasis. Hence, the mitochondrial steady state is essential for cellular function [5-7]. ...
Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): a prospective cohort study
1
2017
... Owing to the epidemiological shift in the 20th century, infectious disease-related death and disability have decreased; however, they have increased rapidly owing to noninfectious diseases including cardiovascular disease (CVD), which have some of the highest mortality and morbidity rates. After the 1930s, mortalities due to CVD exceeded those due to tumors, tuberculosis, and pneumonia. CVD became a leading cause of mortality in developed countries [1]. CVD includes heart and vascular diseases and is a major public health concern and cause of mortality [2]. The prevalence of CVD has increased notwithstanding improvements in treatment and management methods. Age-standardized mortality of CVD has decreased by 14.5% over the past two decades [3]. Nonetheless, with an increase in the incidence of cardiovascular disease and changes in the disease spectrum, the incidence of CVD and CVD-associated mortality are expected to increase worldwide in future decades. Therefore, prevention and treatment of cardiovascular disease is extremely urgent [4]. In CVD, the final and the most prominent phenomenon is cell death caused by harmful stimuli and aging. Mitochondria provide energy for cellular metabolism and are involved in cell death. They participate in cell proliferation, apoptosis, signal transduction, and calcium homeostasis. Hence, the mitochondrial steady state is essential for cellular function [5-7]. ...
Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the Global Burden of Disease Study 2016
1
2017
... Owing to the epidemiological shift in the 20th century, infectious disease-related death and disability have decreased; however, they have increased rapidly owing to noninfectious diseases including cardiovascular disease (CVD), which have some of the highest mortality and morbidity rates. After the 1930s, mortalities due to CVD exceeded those due to tumors, tuberculosis, and pneumonia. CVD became a leading cause of mortality in developed countries [1]. CVD includes heart and vascular diseases and is a major public health concern and cause of mortality [2]. The prevalence of CVD has increased notwithstanding improvements in treatment and management methods. Age-standardized mortality of CVD has decreased by 14.5% over the past two decades [3]. Nonetheless, with an increase in the incidence of cardiovascular disease and changes in the disease spectrum, the incidence of CVD and CVD-associated mortality are expected to increase worldwide in future decades. Therefore, prevention and treatment of cardiovascular disease is extremely urgent [4]. In CVD, the final and the most prominent phenomenon is cell death caused by harmful stimuli and aging. Mitochondria provide energy for cellular metabolism and are involved in cell death. They participate in cell proliferation, apoptosis, signal transduction, and calcium homeostasis. Hence, the mitochondrial steady state is essential for cellular function [5-7]. ...
Cardiovascular risk and events in 17 low-, middle-, and high-income countries
1
2014
... Owing to the epidemiological shift in the 20th century, infectious disease-related death and disability have decreased; however, they have increased rapidly owing to noninfectious diseases including cardiovascular disease (CVD), which have some of the highest mortality and morbidity rates. After the 1930s, mortalities due to CVD exceeded those due to tumors, tuberculosis, and pneumonia. CVD became a leading cause of mortality in developed countries [1]. CVD includes heart and vascular diseases and is a major public health concern and cause of mortality [2]. The prevalence of CVD has increased notwithstanding improvements in treatment and management methods. Age-standardized mortality of CVD has decreased by 14.5% over the past two decades [3]. Nonetheless, with an increase in the incidence of cardiovascular disease and changes in the disease spectrum, the incidence of CVD and CVD-associated mortality are expected to increase worldwide in future decades. Therefore, prevention and treatment of cardiovascular disease is extremely urgent [4]. In CVD, the final and the most prominent phenomenon is cell death caused by harmful stimuli and aging. Mitochondria provide energy for cellular metabolism and are involved in cell death. They participate in cell proliferation, apoptosis, signal transduction, and calcium homeostasis. Hence, the mitochondrial steady state is essential for cellular function [5-7]. ...
Targeting mitochondria for cardiovascular disorders: therapeutic potential and obstacles
1
2019
... Owing to the epidemiological shift in the 20th century, infectious disease-related death and disability have decreased; however, they have increased rapidly owing to noninfectious diseases including cardiovascular disease (CVD), which have some of the highest mortality and morbidity rates. After the 1930s, mortalities due to CVD exceeded those due to tumors, tuberculosis, and pneumonia. CVD became a leading cause of mortality in developed countries [1]. CVD includes heart and vascular diseases and is a major public health concern and cause of mortality [2]. The prevalence of CVD has increased notwithstanding improvements in treatment and management methods. Age-standardized mortality of CVD has decreased by 14.5% over the past two decades [3]. Nonetheless, with an increase in the incidence of cardiovascular disease and changes in the disease spectrum, the incidence of CVD and CVD-associated mortality are expected to increase worldwide in future decades. Therefore, prevention and treatment of cardiovascular disease is extremely urgent [4]. In CVD, the final and the most prominent phenomenon is cell death caused by harmful stimuli and aging. Mitochondria provide energy for cellular metabolism and are involved in cell death. They participate in cell proliferation, apoptosis, signal transduction, and calcium homeostasis. Hence, the mitochondrial steady state is essential for cellular function [5-7]. ...
Mitochondrial control of cellular life, stress, and death
0
2012
Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology
1
2017
... Owing to the epidemiological shift in the 20th century, infectious disease-related death and disability have decreased; however, they have increased rapidly owing to noninfectious diseases including cardiovascular disease (CVD), which have some of the highest mortality and morbidity rates. After the 1930s, mortalities due to CVD exceeded those due to tumors, tuberculosis, and pneumonia. CVD became a leading cause of mortality in developed countries [1]. CVD includes heart and vascular diseases and is a major public health concern and cause of mortality [2]. The prevalence of CVD has increased notwithstanding improvements in treatment and management methods. Age-standardized mortality of CVD has decreased by 14.5% over the past two decades [3]. Nonetheless, with an increase in the incidence of cardiovascular disease and changes in the disease spectrum, the incidence of CVD and CVD-associated mortality are expected to increase worldwide in future decades. Therefore, prevention and treatment of cardiovascular disease is extremely urgent [4]. In CVD, the final and the most prominent phenomenon is cell death caused by harmful stimuli and aging. Mitochondria provide energy for cellular metabolism and are involved in cell death. They participate in cell proliferation, apoptosis, signal transduction, and calcium homeostasis. Hence, the mitochondrial steady state is essential for cellular function [5-7]. ...
Mitophagy as a Protective Mechanism against Myocardial Stress
1
2017
... Mitochondria are the most abundant organelles in cardiomyocytes and are very important for the maintenance of normal cell function. Mitophagy is selective autophagy wherein cells eliminate damaged or dysfunctional mitochondria, which is essential for the regulation of mitochondrial homeostasis [8-10]. Mitophagy was first proposed by Lemasters et al. in 2005. Since then, numerous diseases have been associated with mitophagy [11]. Recent studies revealed that mitophagy contributes to the pathophysiology of CVD. Mitophagy regulates cardiovascular activity through several pathways [12-22]. The present review discusses mitophagy and its underlying mechanisms, methods to assess mitophagy and its involvements in CVD. ...
Aging and Autophagy in the Heart
0
2016
Does Autophagy Mediate Cardiac Myocyte Death During Stress?
1
2016
... Mitochondria are the most abundant organelles in cardiomyocytes and are very important for the maintenance of normal cell function. Mitophagy is selective autophagy wherein cells eliminate damaged or dysfunctional mitochondria, which is essential for the regulation of mitochondrial homeostasis [8-10]. Mitophagy was first proposed by Lemasters et al. in 2005. Since then, numerous diseases have been associated with mitophagy [11]. Recent studies revealed that mitophagy contributes to the pathophysiology of CVD. Mitophagy regulates cardiovascular activity through several pathways [12-22]. The present review discusses mitophagy and its underlying mechanisms, methods to assess mitophagy and its involvements in CVD. ...
Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging
2
2005
... Mitochondria are the most abundant organelles in cardiomyocytes and are very important for the maintenance of normal cell function. Mitophagy is selective autophagy wherein cells eliminate damaged or dysfunctional mitochondria, which is essential for the regulation of mitochondrial homeostasis [8-10]. Mitophagy was first proposed by Lemasters et al. in 2005. Since then, numerous diseases have been associated with mitophagy [11]. Recent studies revealed that mitophagy contributes to the pathophysiology of CVD. Mitophagy regulates cardiovascular activity through several pathways [12-22]. The present review discusses mitophagy and its underlying mechanisms, methods to assess mitophagy and its involvements in CVD. ...
... Mitochondria regulate calcium steady state, signal transmission between organelles, and oxidative phosphorylation for ATP biosynthesis. Mitochondrial DNA (mtDNA) is present in the matrix or membrane and does not combine with histones. Therefore, mtDNAs are relatively vulnerable to reactive oxygen species (ROS) and influenced by mutations. Mitochondria are more susceptible to oxidative damage than other organelles [23-25]. Dysfunctional and damaged mitochondria generate excess ROS, which damage proteins, membrane lipids, nucleic acids, adjacent mitochondria and release proapoptotic proteins into the cytosol, eventually causing cell death [25-28]. Hence, prompt and appropriate elimination of damaged or aging mitochondria is vital for normal cell growth. This process is executed by mitophagy [11]. ...
Mitochondrial biogenesis and dynamics in the developing and diseased heart
1
2015
... Mitochondria are the most abundant organelles in cardiomyocytes and are very important for the maintenance of normal cell function. Mitophagy is selective autophagy wherein cells eliminate damaged or dysfunctional mitochondria, which is essential for the regulation of mitochondrial homeostasis [8-10]. Mitophagy was first proposed by Lemasters et al. in 2005. Since then, numerous diseases have been associated with mitophagy [11]. Recent studies revealed that mitophagy contributes to the pathophysiology of CVD. Mitophagy regulates cardiovascular activity through several pathways [12-22]. The present review discusses mitophagy and its underlying mechanisms, methods to assess mitophagy and its involvements in CVD. ...
Mitochondrial Metabolism in Aging Heart
0
2016
Mitochondrial quality control in the diabetic heart
1
2016
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Mending a broken heart: the role of mitophagy in cardioprotection
1
2015
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart
4
2015
... Autophagy involves the degradation of the protoplasm by lysosomes, resulting in macrophage-, microphage-, and chaperone-mediated autophagy. Selective autophagy occurs in response to nutrient deficiency, whereas nonspecific autophagy is caused by cell aging or a response to damaged cell components. Mitophagy is a selective autophagy pathway involving the elimination of abnormal mitochondria and maintains mitochondrial homeostasis [16, 29-32]. ...
... Mitophagy is divided into three steps: formation of phagocytic vesicles, recognition of damaged mitochondrial components and compartmentation into mitochondrial phagocytic vesicles, and fusion of mitochondrial phagocytic vesicles and lysosomes into mitolysosomes [16, 33, 34](Fig. 1). The mitophagy pathway was first identified in yeast in 2004. Mitochondrial outer membrane protein autophagy-related gene 32 (Atg32) recruits core autophagy molecules to the mitochondria for selective clearance [35-37]; however, no homologs have been reported in the human genome thus far. The subsequent section summarizes the mechanisms underlying mitophagy in accordance with the three aforementioned stages. ...
... In mammalian cells, PINK1/Parkin-mediated mitophagy is the most extensively characterized mitophagic phenomenon thus far [43]. PINK1 is a serine/threonine kinase with an N-terminal mitochondrial targeting signal [44]. In intact mitochondria, PINK1 is recruited to the mitochondria by translational enzymes on the outer mitochondrial membrane (OMM) and the endometrial complex, anchored within the mitochondrial endometrium, and continuously degraded by protease and peptidase. During mitochondrial depolarization, however, translocation of PINK1 across to the inner mitochondrial membrane (IMM) is inhibited. PINK1 is not degraded but aggregates at the outer membrane [16, 42]. ...
... LC3 receptors BNIP3/Nix and FUNDC1 have been extensively characterized in cardiovascular studies. BCL2-like13 (Bcl2L13), autophagy/Beclin 1 regulator 1 (AMBRA1), and cardio phospholipids serve as LC3 receptors during mitophagy [16, 42, 58]. They localize at the OMM thus eliminating the need for mitophagic receptors to mark the damaged mitochondria. Rather, they directly interact with LC3 via the LIR and identify and engulf the damaged mitochondria [42, 48, 50]. ...
Mitochondrial quality control: The role of mitophagy in aging
1
2018
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
How mitochondrial dynamism orchestrates mitophagy
0
2015
The Mitochondrial Basis of Aging
2
2016
... In normal cells, mitophagy may have a housekeeping function and renew mitochondrial components. Mitophagy participates in metabolic remodeling and is closely associated with inflammatory response factors. For example, orally administered polyaminospermine in mice enhances mitophagy, inhibits subclinical inflammation, and increases longevity. Changes in the degree of mitophagy modulate stem cell activity and chronic cardiomyocyte inflammation [19, 84]. ...
... Cellular aging has a prominent mitochondrial basis. Mitophagy reportedly mediates various degenerative diseases and contributes to their pathophysiology and treatment. For example, mitophagy is disrupted in the heart of aged individuals and in senescent cells and DOX-treated hearts and cells [19, 86]. ...
Mitochondrial autophagy in cardiomyopathy
0
2016
Autophagy-A key pathway for cardiac health and longevity
0
2018
Current Mechanistic Concepts in Ischemia and Reperfusion Injury
1
2018
... Mitochondria are the most abundant organelles in cardiomyocytes and are very important for the maintenance of normal cell function. Mitophagy is selective autophagy wherein cells eliminate damaged or dysfunctional mitochondria, which is essential for the regulation of mitochondrial homeostasis [8-10]. Mitophagy was first proposed by Lemasters et al. in 2005. Since then, numerous diseases have been associated with mitophagy [11]. Recent studies revealed that mitophagy contributes to the pathophysiology of CVD. Mitophagy regulates cardiovascular activity through several pathways [12-22]. The present review discusses mitophagy and its underlying mechanisms, methods to assess mitophagy and its involvements in CVD. ...
The cardiac mitochondrion: nexus of stress
1
2010
... Mitochondria regulate calcium steady state, signal transmission between organelles, and oxidative phosphorylation for ATP biosynthesis. Mitochondrial DNA (mtDNA) is present in the matrix or membrane and does not combine with histones. Therefore, mtDNAs are relatively vulnerable to reactive oxygen species (ROS) and influenced by mutations. Mitochondria are more susceptible to oxidative damage than other organelles [23-25]. Dysfunctional and damaged mitochondria generate excess ROS, which damage proteins, membrane lipids, nucleic acids, adjacent mitochondria and release proapoptotic proteins into the cytosol, eventually causing cell death [25-28]. Hence, prompt and appropriate elimination of damaged or aging mitochondria is vital for normal cell growth. This process is executed by mitophagy [11]. ...
Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria
0
2010
Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging
2
2005
... Mitochondria regulate calcium steady state, signal transmission between organelles, and oxidative phosphorylation for ATP biosynthesis. Mitochondrial DNA (mtDNA) is present in the matrix or membrane and does not combine with histones. Therefore, mtDNAs are relatively vulnerable to reactive oxygen species (ROS) and influenced by mutations. Mitochondria are more susceptible to oxidative damage than other organelles [23-25]. Dysfunctional and damaged mitochondria generate excess ROS, which damage proteins, membrane lipids, nucleic acids, adjacent mitochondria and release proapoptotic proteins into the cytosol, eventually causing cell death [25-28]. Hence, prompt and appropriate elimination of damaged or aging mitochondria is vital for normal cell growth. This process is executed by mitophagy [11]. ...
... ]. Dysfunctional and damaged mitochondria generate excess ROS, which damage proteins, membrane lipids, nucleic acids, adjacent mitochondria and release proapoptotic proteins into the cytosol, eventually causing cell death [25-28]. Hence, prompt and appropriate elimination of damaged or aging mitochondria is vital for normal cell growth. This process is executed by mitophagy [11]. ...
Mitochondria and cardiovascular aging
1
2012
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine
0
2005
Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction
3
2013
... Mitochondria regulate calcium steady state, signal transmission between organelles, and oxidative phosphorylation for ATP biosynthesis. Mitochondrial DNA (mtDNA) is present in the matrix or membrane and does not combine with histones. Therefore, mtDNAs are relatively vulnerable to reactive oxygen species (ROS) and influenced by mutations. Mitochondria are more susceptible to oxidative damage than other organelles [23-25]. Dysfunctional and damaged mitochondria generate excess ROS, which damage proteins, membrane lipids, nucleic acids, adjacent mitochondria and release proapoptotic proteins into the cytosol, eventually causing cell death [25-28]. Hence, prompt and appropriate elimination of damaged or aging mitochondria is vital for normal cell growth. This process is executed by mitophagy [11]. ...
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
Mitochondrial Quality Control and Disease: Insights into Ischemia-Reperfusion Injury
2
2018
... Autophagy involves the degradation of the protoplasm by lysosomes, resulting in macrophage-, microphage-, and chaperone-mediated autophagy. Selective autophagy occurs in response to nutrient deficiency, whereas nonspecific autophagy is caused by cell aging or a response to damaged cell components. Mitophagy is a selective autophagy pathway involving the elimination of abnormal mitochondria and maintains mitochondrial homeostasis [16, 29-32]. ...
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
The critical roles of mitophagy in cerebral ischemia
1
2016
... Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163]. ...
Chaperone-mediated autophagy: roles in disease and aging
0
2014
Turnover of organelles by autophagy in yeast
1
2009
... Autophagy involves the degradation of the protoplasm by lysosomes, resulting in macrophage-, microphage-, and chaperone-mediated autophagy. Selective autophagy occurs in response to nutrient deficiency, whereas nonspecific autophagy is caused by cell aging or a response to damaged cell components. Mitophagy is a selective autophagy pathway involving the elimination of abnormal mitochondria and maintains mitochondrial homeostasis [16, 29-32]. ...
Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy
4
2016
... Mitophagy is divided into three steps: formation of phagocytic vesicles, recognition of damaged mitochondrial components and compartmentation into mitochondrial phagocytic vesicles, and fusion of mitochondrial phagocytic vesicles and lysosomes into mitolysosomes [16, 33, 34](Fig. 1). The mitophagy pathway was first identified in yeast in 2004. Mitochondrial outer membrane protein autophagy-related gene 32 (Atg32) recruits core autophagy molecules to the mitochondria for selective clearance [35-37]; however, no homologs have been reported in the human genome thus far. The subsequent section summarizes the mechanisms underlying mitophagy in accordance with the three aforementioned stages. ...
... Phagocytic vesicles are isolated membranous structures in the endoplasmic reticulum (ER) of mammalian cells. The formation of phagocytic vesicles is regulated by an evolutionarily conserved protein (Atg) system [35, 37-39]. Atg6 recruits other Atg proteins to the isolation membrane, thus warranting the Atg5-Atg12 and LC3-phosphatidylethanolamine conjugation systems. Covalent conjugation between Atg12 and Atg5 resembles ubiquitination, wherein Atg12 is initially activated by Atg7, and then transferred to Atg10. Atg12 is then covalently conjugated with Atg5 at a lysine residue and Atg10 is released. Finally, the Atg12-Atg5 complex interacts non-covalently with Atg16, which then initiates the elongation of the membrane by recruiting LC3-phosphatidylethanolamine [39]. LC3 is a surface molecule on phagocytic vesicles. Phagocytic vesicles are formed by combining LC3-I with phosphatidylethanolamine to produce lipid LC3-II [33, 40, 41]. ...
... Mitophagy is a type of selective organellar autophagy in mammalian cells. Identification of phagocytic vesicles and their combination with damaged mitochondria have received increasing attention in studies on mitophagy [33, 42]. Damaged mitochondria are recognized by LC3 adapters or LC3 receptors. First, we address the recognition of impaired mitochondria by LC3 adapters. ...
... In summary, the impaired mitochondrial recognition process mediated by the LC3 adapter is divided into the ubiquitin-associated type (PINK1/Parkin) and the ubiquitin-independent type (CHDH). Five LC3 adapters have been identified including OPTN, p62/SQSTM1, NBR1, NDP52, and TAX1BP, which mediate the recognition and phagocytosis of damaged mitochondria by joining LC3-coated phagocytic vesicles to label damaged mitochondria [33, 42]. Further, we discuss the LC3 reporter-mediated recognition of impaired mitochondria. ...
Mitophagy Transcriptome: Mechanistic Insights into Polyphenol-Mediated Mitophagy
1
2017
... Mitophagy is divided into three steps: formation of phagocytic vesicles, recognition of damaged mitochondrial components and compartmentation into mitochondrial phagocytic vesicles, and fusion of mitochondrial phagocytic vesicles and lysosomes into mitolysosomes [16, 33, 34](Fig. 1). The mitophagy pathway was first identified in yeast in 2004. Mitochondrial outer membrane protein autophagy-related gene 32 (Atg32) recruits core autophagy molecules to the mitochondria for selective clearance [35-37]; however, no homologs have been reported in the human genome thus far. The subsequent section summarizes the mechanisms underlying mitophagy in accordance with the three aforementioned stages. ...
A landmark protein essential for mitophagy: Atg32 recruits the autophagic machinery to mitochondria
2
2009
... Mitophagy is divided into three steps: formation of phagocytic vesicles, recognition of damaged mitochondrial components and compartmentation into mitochondrial phagocytic vesicles, and fusion of mitochondrial phagocytic vesicles and lysosomes into mitolysosomes [16, 33, 34](Fig. 1). The mitophagy pathway was first identified in yeast in 2004. Mitochondrial outer membrane protein autophagy-related gene 32 (Atg32) recruits core autophagy molecules to the mitochondria for selective clearance [35-37]; however, no homologs have been reported in the human genome thus far. The subsequent section summarizes the mechanisms underlying mitophagy in accordance with the three aforementioned stages. ...
... Phagocytic vesicles are isolated membranous structures in the endoplasmic reticulum (ER) of mammalian cells. The formation of phagocytic vesicles is regulated by an evolutionarily conserved protein (Atg) system [35, 37-39]. Atg6 recruits other Atg proteins to the isolation membrane, thus warranting the Atg5-Atg12 and LC3-phosphatidylethanolamine conjugation systems. Covalent conjugation between Atg12 and Atg5 resembles ubiquitination, wherein Atg12 is initially activated by Atg7, and then transferred to Atg10. Atg12 is then covalently conjugated with Atg5 at a lysine residue and Atg10 is released. Finally, the Atg12-Atg5 complex interacts non-covalently with Atg16, which then initiates the elongation of the membrane by recruiting LC3-phosphatidylethanolamine [39]. LC3 is a surface molecule on phagocytic vesicles. Phagocytic vesicles are formed by combining LC3-I with phosphatidylethanolamine to produce lipid LC3-II [33, 40, 41]. ...
Uth1p is involved in the autophagic degradation of mitochondria
0
2004
Atg32 is a mitochondrial protein that confers selectivity during mitophagy
2
2009
... Mitophagy is divided into three steps: formation of phagocytic vesicles, recognition of damaged mitochondrial components and compartmentation into mitochondrial phagocytic vesicles, and fusion of mitochondrial phagocytic vesicles and lysosomes into mitolysosomes [16, 33, 34](Fig. 1). The mitophagy pathway was first identified in yeast in 2004. Mitochondrial outer membrane protein autophagy-related gene 32 (Atg32) recruits core autophagy molecules to the mitochondria for selective clearance [35-37]; however, no homologs have been reported in the human genome thus far. The subsequent section summarizes the mechanisms underlying mitophagy in accordance with the three aforementioned stages. ...
... Phagocytic vesicles are isolated membranous structures in the endoplasmic reticulum (ER) of mammalian cells. The formation of phagocytic vesicles is regulated by an evolutionarily conserved protein (Atg) system [35, 37-39]. Atg6 recruits other Atg proteins to the isolation membrane, thus warranting the Atg5-Atg12 and LC3-phosphatidylethanolamine conjugation systems. Covalent conjugation between Atg12 and Atg5 resembles ubiquitination, wherein Atg12 is initially activated by Atg7, and then transferred to Atg10. Atg12 is then covalently conjugated with Atg5 at a lysine residue and Atg10 is released. Finally, the Atg12-Atg5 complex interacts non-covalently with Atg16, which then initiates the elongation of the membrane by recruiting LC3-phosphatidylethanolamine [39]. LC3 is a surface molecule on phagocytic vesicles. Phagocytic vesicles are formed by combining LC3-I with phosphatidylethanolamine to produce lipid LC3-II [33, 40, 41]. ...
Studies on the mechanisms of autophagy: formation of the autophagic vacuole
0
1990
Recycle or die: the role of autophagy in cardioprotection
2
2008
... Phagocytic vesicles are isolated membranous structures in the endoplasmic reticulum (ER) of mammalian cells. The formation of phagocytic vesicles is regulated by an evolutionarily conserved protein (Atg) system [35, 37-39]. Atg6 recruits other Atg proteins to the isolation membrane, thus warranting the Atg5-Atg12 and LC3-phosphatidylethanolamine conjugation systems. Covalent conjugation between Atg12 and Atg5 resembles ubiquitination, wherein Atg12 is initially activated by Atg7, and then transferred to Atg10. Atg12 is then covalently conjugated with Atg5 at a lysine residue and Atg10 is released. Finally, the Atg12-Atg5 complex interacts non-covalently with Atg16, which then initiates the elongation of the membrane by recruiting LC3-phosphatidylethanolamine [39]. LC3 is a surface molecule on phagocytic vesicles. Phagocytic vesicles are formed by combining LC3-I with phosphatidylethanolamine to produce lipid LC3-II [33, 40, 41]. ...
... ]. Atg6 recruits other Atg proteins to the isolation membrane, thus warranting the Atg5-Atg12 and LC3-phosphatidylethanolamine conjugation systems. Covalent conjugation between Atg12 and Atg5 resembles ubiquitination, wherein Atg12 is initially activated by Atg7, and then transferred to Atg10. Atg12 is then covalently conjugated with Atg5 at a lysine residue and Atg10 is released. Finally, the Atg12-Atg5 complex interacts non-covalently with Atg16, which then initiates the elongation of the membrane by recruiting LC3-phosphatidylethanolamine [39]. LC3 is a surface molecule on phagocytic vesicles. Phagocytic vesicles are formed by combining LC3-I with phosphatidylethanolamine to produce lipid LC3-II [33, 40, 41]. ...
Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3)
1
2014
... Phagocytic vesicles are isolated membranous structures in the endoplasmic reticulum (ER) of mammalian cells. The formation of phagocytic vesicles is regulated by an evolutionarily conserved protein (Atg) system [35, 37-39]. Atg6 recruits other Atg proteins to the isolation membrane, thus warranting the Atg5-Atg12 and LC3-phosphatidylethanolamine conjugation systems. Covalent conjugation between Atg12 and Atg5 resembles ubiquitination, wherein Atg12 is initially activated by Atg7, and then transferred to Atg10. Atg12 is then covalently conjugated with Atg5 at a lysine residue and Atg10 is released. Finally, the Atg12-Atg5 complex interacts non-covalently with Atg16, which then initiates the elongation of the membrane by recruiting LC3-phosphatidylethanolamine [39]. LC3 is a surface molecule on phagocytic vesicles. Phagocytic vesicles are formed by combining LC3-I with phosphatidylethanolamine to produce lipid LC3-II [33, 40, 41]. ...
LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing
2
2000
... Phagocytic vesicles are isolated membranous structures in the endoplasmic reticulum (ER) of mammalian cells. The formation of phagocytic vesicles is regulated by an evolutionarily conserved protein (Atg) system [35, 37-39]. Atg6 recruits other Atg proteins to the isolation membrane, thus warranting the Atg5-Atg12 and LC3-phosphatidylethanolamine conjugation systems. Covalent conjugation between Atg12 and Atg5 resembles ubiquitination, wherein Atg12 is initially activated by Atg7, and then transferred to Atg10. Atg12 is then covalently conjugated with Atg5 at a lysine residue and Atg10 is released. Finally, the Atg12-Atg5 complex interacts non-covalently with Atg16, which then initiates the elongation of the membrane by recruiting LC3-phosphatidylethanolamine [39]. LC3 is a surface molecule on phagocytic vesicles. Phagocytic vesicles are formed by combining LC3-I with phosphatidylethanolamine to produce lipid LC3-II [33, 40, 41]. ...
... A commonly used method to detect autophagy proteins is western blotting. Autophagy can be detected on the basis of the ratio of the LC3B II/I type marker protein and the extent of P62 degradation [61]. Mitophagy is accompanied by the degradation of several mitochondrial proteins; hence, it may also be evaluated by measuring their levels. Mitochondrial degradation occurs through the autophagy pathway, and cytosolic LC3 protein is converted from type I to type II via conjugation with phosphatidylethanolamine. Therefore, LC3 detection is also a potential method of detecting autophagy. The use of autophagy inhibitors including Bafilomycin A1 (Baf A1) and hydroxychloroquine (HCQ) is also a prominent method of detecting autophagy [41, 62]. Recent studies have reported that the OMM, IMM, and matrix proteins are degraded via mitophagy, whereas certain proteins regulating mitochondrial division and migration are degraded by proteasomes or ATP-dependent protease pathways. TIM23, TOM20, Hsp60, and electron transport chain (ETC)-related proteins are commonly used as mitophagy markers [63]. P62 can be recruited individually, since it is highly ubiquitinated after mitochondrial damage. P62 is involved in numerous signal transduction pathways and its function is complex. Therefore, the role of P62 in mitophagy detection is controversial [64, 65]. ...
A Molecular Approach to Mitophagy and Mitochondrial Dynamics
6
2018
... Mitophagy is a type of selective organellar autophagy in mammalian cells. Identification of phagocytic vesicles and their combination with damaged mitochondria have received increasing attention in studies on mitophagy [33, 42]. Damaged mitochondria are recognized by LC3 adapters or LC3 receptors. First, we address the recognition of impaired mitochondria by LC3 adapters. ...
... In mammalian cells, PINK1/Parkin-mediated mitophagy is the most extensively characterized mitophagic phenomenon thus far [43]. PINK1 is a serine/threonine kinase with an N-terminal mitochondrial targeting signal [44]. In intact mitochondria, PINK1 is recruited to the mitochondria by translational enzymes on the outer mitochondrial membrane (OMM) and the endometrial complex, anchored within the mitochondrial endometrium, and continuously degraded by protease and peptidase. During mitochondrial depolarization, however, translocation of PINK1 across to the inner mitochondrial membrane (IMM) is inhibited. PINK1 is not degraded but aggregates at the outer membrane [16, 42]. ...
... CHDH oxidizes choline to betaine aldehyde in the mitochondria. In normal mitochondria, CHDH is unanchored at both the OMM and IMM. In depolarized mitochondria, CHDH accumulates on the OMM and its FAD/NAD-binding domain 1 interacts with the Phox and Bem1 domains in cytoplasmic p62/SQSTM1, thus recruiting p62/SQSTM1 at damaged mitochondria and binding with LC3 [42, 50]. ...
... In summary, the impaired mitochondrial recognition process mediated by the LC3 adapter is divided into the ubiquitin-associated type (PINK1/Parkin) and the ubiquitin-independent type (CHDH). Five LC3 adapters have been identified including OPTN, p62/SQSTM1, NBR1, NDP52, and TAX1BP, which mediate the recognition and phagocytosis of damaged mitochondria by joining LC3-coated phagocytic vesicles to label damaged mitochondria [33, 42]. Further, we discuss the LC3 reporter-mediated recognition of impaired mitochondria. ...
... LC3 receptors BNIP3/Nix and FUNDC1 have been extensively characterized in cardiovascular studies. BCL2-like13 (Bcl2L13), autophagy/Beclin 1 regulator 1 (AMBRA1), and cardio phospholipids serve as LC3 receptors during mitophagy [16, 42, 58]. They localize at the OMM thus eliminating the need for mitophagic receptors to mark the damaged mitochondria. Rather, they directly interact with LC3 via the LIR and identify and engulf the damaged mitochondria [42, 48, 50]. ...
... ]. They localize at the OMM thus eliminating the need for mitophagic receptors to mark the damaged mitochondria. Rather, they directly interact with LC3 via the LIR and identify and engulf the damaged mitochondria [42, 48, 50]. ...
Mechanisms of mitophagy
2
2011
... In mammalian cells, PINK1/Parkin-mediated mitophagy is the most extensively characterized mitophagic phenomenon thus far [43]. PINK1 is a serine/threonine kinase with an N-terminal mitochondrial targeting signal [44]. In intact mitochondria, PINK1 is recruited to the mitochondria by translational enzymes on the outer mitochondrial membrane (OMM) and the endometrial complex, anchored within the mitochondrial endometrium, and continuously degraded by protease and peptidase. During mitochondrial depolarization, however, translocation of PINK1 across to the inner mitochondrial membrane (IMM) is inhibited. PINK1 is not degraded but aggregates at the outer membrane [16, 42]. ...
... PINK1 utilizes its own ubiquitin-like domain to function like ubiquitin. Activated PINK1 phosphorylates Parkin (a cytoplasmic E3 ubiquitin ligase) at T175, thus activating Parkin, which in turn aggregates in the OMM. However, activated PINK1 also phosphorylates Mfn2, which can then serve as a Parkin receptor in cardiomyocyte mitochondria. This process also induces Parkin aggregation on the OMM [43, 45-47]. Phospho-parkin helps generate ubiquitin chains on the OMM proteins through polyubiquitination, thus amplifying the signal, and these ubiquitin chains serve as LC3 adapter recognition sites, which bind to LC3 and initiate mitophagy [48, 49]. ...
Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL
1
2010
... In mammalian cells, PINK1/Parkin-mediated mitophagy is the most extensively characterized mitophagic phenomenon thus far [43]. PINK1 is a serine/threonine kinase with an N-terminal mitochondrial targeting signal [44]. In intact mitochondria, PINK1 is recruited to the mitochondria by translational enzymes on the outer mitochondrial membrane (OMM) and the endometrial complex, anchored within the mitochondrial endometrium, and continuously degraded by protease and peptidase. During mitochondrial depolarization, however, translocation of PINK1 across to the inner mitochondrial membrane (IMM) is inhibited. PINK1 is not degraded but aggregates at the outer membrane [16, 42]. ...
PINK1 is selectively stabilized on impaired mitochondria to activate Parkin
1
2010
... PINK1 utilizes its own ubiquitin-like domain to function like ubiquitin. Activated PINK1 phosphorylates Parkin (a cytoplasmic E3 ubiquitin ligase) at T175, thus activating Parkin, which in turn aggregates in the OMM. However, activated PINK1 also phosphorylates Mfn2, which can then serve as a Parkin receptor in cardiomyocyte mitochondria. This process also induces Parkin aggregation on the OMM [43, 45-47]. Phospho-parkin helps generate ubiquitin chains on the OMM proteins through polyubiquitination, thus amplifying the signal, and these ubiquitin chains serve as LC3 adapter recognition sites, which bind to LC3 and initiate mitophagy [48, 49]. ...
Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment
0
2012
Autoregulation of Parkin activity through its ubiquitin-like domain
1
2011
... PINK1 utilizes its own ubiquitin-like domain to function like ubiquitin. Activated PINK1 phosphorylates Parkin (a cytoplasmic E3 ubiquitin ligase) at T175, thus activating Parkin, which in turn aggregates in the OMM. However, activated PINK1 also phosphorylates Mfn2, which can then serve as a Parkin receptor in cardiomyocyte mitochondria. This process also induces Parkin aggregation on the OMM [43, 45-47]. Phospho-parkin helps generate ubiquitin chains on the OMM proteins through polyubiquitination, thus amplifying the signal, and these ubiquitin chains serve as LC3 adapter recognition sites, which bind to LC3 and initiate mitophagy [48, 49]. ...
The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy
3
2015
... PINK1 utilizes its own ubiquitin-like domain to function like ubiquitin. Activated PINK1 phosphorylates Parkin (a cytoplasmic E3 ubiquitin ligase) at T175, thus activating Parkin, which in turn aggregates in the OMM. However, activated PINK1 also phosphorylates Mfn2, which can then serve as a Parkin receptor in cardiomyocyte mitochondria. This process also induces Parkin aggregation on the OMM [43, 45-47]. Phospho-parkin helps generate ubiquitin chains on the OMM proteins through polyubiquitination, thus amplifying the signal, and these ubiquitin chains serve as LC3 adapter recognition sites, which bind to LC3 and initiate mitophagy [48, 49]. ...
... PINK1 may also be recruited in a Parkin-independent manner. PINK1 phosphorylates the mitochondrial ubiquitin on OMM at S65. LC3 adapters such as optineurin and NDP52 recognize the damaged mitochondria and ubiquitin phosphorylated at S65 through the ubiquitin-binding region [48]. ...
... LC3 receptors BNIP3/Nix and FUNDC1 have been extensively characterized in cardiovascular studies. BCL2-like13 (Bcl2L13), autophagy/Beclin 1 regulator 1 (AMBRA1), and cardio phospholipids serve as LC3 receptors during mitophagy [16, 42, 58]. They localize at the OMM thus eliminating the need for mitophagic receptors to mark the damaged mitochondria. Rather, they directly interact with LC3 via the LIR and identify and engulf the damaged mitochondria [42, 48, 50]. ...
Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy
1
2011
... PINK1 utilizes its own ubiquitin-like domain to function like ubiquitin. Activated PINK1 phosphorylates Parkin (a cytoplasmic E3 ubiquitin ligase) at T175, thus activating Parkin, which in turn aggregates in the OMM. However, activated PINK1 also phosphorylates Mfn2, which can then serve as a Parkin receptor in cardiomyocyte mitochondria. This process also induces Parkin aggregation on the OMM [43, 45-47]. Phospho-parkin helps generate ubiquitin chains on the OMM proteins through polyubiquitination, thus amplifying the signal, and these ubiquitin chains serve as LC3 adapter recognition sites, which bind to LC3 and initiate mitophagy [48, 49]. ...
Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy
2
2014
... CHDH oxidizes choline to betaine aldehyde in the mitochondria. In normal mitochondria, CHDH is unanchored at both the OMM and IMM. In depolarized mitochondria, CHDH accumulates on the OMM and its FAD/NAD-binding domain 1 interacts with the Phox and Bem1 domains in cytoplasmic p62/SQSTM1, thus recruiting p62/SQSTM1 at damaged mitochondria and binding with LC3 [42, 50]. ...
... LC3 receptors BNIP3/Nix and FUNDC1 have been extensively characterized in cardiovascular studies. BCL2-like13 (Bcl2L13), autophagy/Beclin 1 regulator 1 (AMBRA1), and cardio phospholipids serve as LC3 receptors during mitophagy [16, 42, 58]. They localize at the OMM thus eliminating the need for mitophagic receptors to mark the damaged mitochondria. Rather, they directly interact with LC3 via the LIR and identify and engulf the damaged mitochondria [42, 48, 50]. ...
Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy
1
2009
... Nix and BNIP3 are OMM proteins that interact with LC3 or its homologs through the LC3-interacting region (LIR) to form mitophagy receptors. BNIP3 and Nix are hypoxia-induced tail-anchored proteins that bind to the OMM via the carboxyl transmembrane domain. The N-terminal extends into the cytoplasm and is linked to LC3-related molecules[51-53]. BNIP3-dependent mitophagy requires drp1 to induce mitochondrial division. Inhibition of this process or drp1 knockout blocks hypoxia-induced mitophagy. Nix protein levels increase after erythrocyte maturation. Nix knockout inhibits the loss of the mitochondrial membrane potential and the isolation of mitochondrial autophagosomes. Nix-dependent mitophagy is induced by the loss of membrane potential caused by carbonyl cyanide m-chlorophenylhydrazone treatment. Therefore, Nix-mediated loss of membrane proteins is important in mitophagy [54]. ...
Nix is a selective autophagy receptor for mitochondrial clearance
0
2010
Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy
1
2012
... Nix and BNIP3 are OMM proteins that interact with LC3 or its homologs through the LC3-interacting region (LIR) to form mitophagy receptors. BNIP3 and Nix are hypoxia-induced tail-anchored proteins that bind to the OMM via the carboxyl transmembrane domain. The N-terminal extends into the cytoplasm and is linked to LC3-related molecules[51-53]. BNIP3-dependent mitophagy requires drp1 to induce mitochondrial division. Inhibition of this process or drp1 knockout blocks hypoxia-induced mitophagy. Nix protein levels increase after erythrocyte maturation. Nix knockout inhibits the loss of the mitochondrial membrane potential and the isolation of mitochondrial autophagosomes. Nix-dependent mitophagy is induced by the loss of membrane potential caused by carbonyl cyanide m-chlorophenylhydrazone treatment. Therefore, Nix-mediated loss of membrane proteins is important in mitophagy [54]. ...
Essential role for Nix in autophagic maturation of erythroid cells
1
2008
... Nix and BNIP3 are OMM proteins that interact with LC3 or its homologs through the LC3-interacting region (LIR) to form mitophagy receptors. BNIP3 and Nix are hypoxia-induced tail-anchored proteins that bind to the OMM via the carboxyl transmembrane domain. The N-terminal extends into the cytoplasm and is linked to LC3-related molecules[51-53]. BNIP3-dependent mitophagy requires drp1 to induce mitochondrial division. Inhibition of this process or drp1 knockout blocks hypoxia-induced mitophagy. Nix protein levels increase after erythrocyte maturation. Nix knockout inhibits the loss of the mitochondrial membrane potential and the isolation of mitochondrial autophagosomes. Nix-dependent mitophagy is induced by the loss of membrane potential caused by carbonyl cyanide m-chlorophenylhydrazone treatment. Therefore, Nix-mediated loss of membrane proteins is important in mitophagy [54]. ...
Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells
1
2012
... FUNDC1 is an OMM protein, which integrates with LC3 under hypoxia via its LIR structure. During starvation, mitophagy does not depend on FUNDC1; therefore, it specifically induces hypoxia [55]. FUNDC1 is expressed under normoxic conditions; however, FUNDC1 and LC3 interact under hypoxic conditions upon dephosphorylation at Y18 [56]. Furthermore, FUNDC1 is a mitochondrial receptor for unc-51-like autophagy activating kinase 1 (ULK1), which in turn is translocated to depolarized mitochondria and FUNDC1 phosphorylated at S17 and promotes mitophagy [56]. Phosphoglycerate mutase family member 5 phosphatase is located at the mitochondria and dephosphorylates FUNDC1 at S13 under hypoxic conditions or upon treatment with carbonyl cyanide p-trifluoromethoxy phenylhydrazone. The latter promotes the interaction of the enzyme with LC3 and induces mitophagy [57]. ...
ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy
2
2014
... FUNDC1 is an OMM protein, which integrates with LC3 under hypoxia via its LIR structure. During starvation, mitophagy does not depend on FUNDC1; therefore, it specifically induces hypoxia [55]. FUNDC1 is expressed under normoxic conditions; however, FUNDC1 and LC3 interact under hypoxic conditions upon dephosphorylation at Y18 [56]. Furthermore, FUNDC1 is a mitochondrial receptor for unc-51-like autophagy activating kinase 1 (ULK1), which in turn is translocated to depolarized mitochondria and FUNDC1 phosphorylated at S17 and promotes mitophagy [56]. Phosphoglycerate mutase family member 5 phosphatase is located at the mitochondria and dephosphorylates FUNDC1 at S13 under hypoxic conditions or upon treatment with carbonyl cyanide p-trifluoromethoxy phenylhydrazone. The latter promotes the interaction of the enzyme with LC3 and induces mitophagy [57]. ...
... ]. Furthermore, FUNDC1 is a mitochondrial receptor for unc-51-like autophagy activating kinase 1 (ULK1), which in turn is translocated to depolarized mitochondria and FUNDC1 phosphorylated at S17 and promotes mitophagy [56]. Phosphoglycerate mutase family member 5 phosphatase is located at the mitochondria and dephosphorylates FUNDC1 at S13 under hypoxic conditions or upon treatment with carbonyl cyanide p-trifluoromethoxy phenylhydrazone. The latter promotes the interaction of the enzyme with LC3 and induces mitophagy [57]. ...
Cost-effectiveness of recombinant tissue-type plasminogen activator within 3 hours of acute ischemic stroke: current evidence
1
2014
... FUNDC1 is an OMM protein, which integrates with LC3 under hypoxia via its LIR structure. During starvation, mitophagy does not depend on FUNDC1; therefore, it specifically induces hypoxia [55]. FUNDC1 is expressed under normoxic conditions; however, FUNDC1 and LC3 interact under hypoxic conditions upon dephosphorylation at Y18 [56]. Furthermore, FUNDC1 is a mitochondrial receptor for unc-51-like autophagy activating kinase 1 (ULK1), which in turn is translocated to depolarized mitochondria and FUNDC1 phosphorylated at S17 and promotes mitophagy [56]. Phosphoglycerate mutase family member 5 phosphatase is located at the mitochondria and dephosphorylates FUNDC1 at S13 under hypoxic conditions or upon treatment with carbonyl cyanide p-trifluoromethoxy phenylhydrazone. The latter promotes the interaction of the enzyme with LC3 and induces mitophagy [57]. ...
AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1
1
2015
... LC3 receptors BNIP3/Nix and FUNDC1 have been extensively characterized in cardiovascular studies. BCL2-like13 (Bcl2L13), autophagy/Beclin 1 regulator 1 (AMBRA1), and cardio phospholipids serve as LC3 receptors during mitophagy [16, 42, 58]. They localize at the OMM thus eliminating the need for mitophagic receptors to mark the damaged mitochondria. Rather, they directly interact with LC3 via the LIR and identify and engulf the damaged mitochondria [42, 48, 50]. ...
Cell- and stimulus-dependent heterogeneity of synaptic vesicle endocytic recycling mechanisms revealed by studies of dynamin 1-null neurons
1
2008
... The most prominent phenomenon in autophagy is the formation of a double-layered membrane around the cytosol and/or organelles. This structure fuses with the lysosome, its contents are released, and a single-layer membrane is formed. Therefore, early and late autophagy are easily distinguished by observing double- and single-membrane structures. Consistent with cell autophagy, early mitophagy potentially results in packaging of the double-membrane structure, which can be readily identified on the basis of its unique structure-mitochondrial crest. During late autophagy, mitochondria are transported to the lysosomes and can be identified from their density or residual structure. TEM is currently the best method to detect mitophagy. It is the "gold standard" method to directly detect autophagy [59, 60]. However, TEM has several limitations. Electron microscopy requires extensive sample preparation. Cell damage potentially resulting from human error interferes with observations. Moreover, mitochondrial structures are difficult to discern during later stages of autophagy. Therefore, this method has low sensitivity. ...
A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation
1
2009
... The most prominent phenomenon in autophagy is the formation of a double-layered membrane around the cytosol and/or organelles. This structure fuses with the lysosome, its contents are released, and a single-layer membrane is formed. Therefore, early and late autophagy are easily distinguished by observing double- and single-membrane structures. Consistent with cell autophagy, early mitophagy potentially results in packaging of the double-membrane structure, which can be readily identified on the basis of its unique structure-mitochondrial crest. During late autophagy, mitochondria are transported to the lysosomes and can be identified from their density or residual structure. TEM is currently the best method to detect mitophagy. It is the "gold standard" method to directly detect autophagy [59, 60]. However, TEM has several limitations. Electron microscopy requires extensive sample preparation. Cell damage potentially resulting from human error interferes with observations. Moreover, mitochondrial structures are difficult to discern during later stages of autophagy. Therefore, this method has low sensitivity. ...
A role for ubiquitin in selective autophagy
1
2009
... A commonly used method to detect autophagy proteins is western blotting. Autophagy can be detected on the basis of the ratio of the LC3B II/I type marker protein and the extent of P62 degradation [61]. Mitophagy is accompanied by the degradation of several mitochondrial proteins; hence, it may also be evaluated by measuring their levels. Mitochondrial degradation occurs through the autophagy pathway, and cytosolic LC3 protein is converted from type I to type II via conjugation with phosphatidylethanolamine. Therefore, LC3 detection is also a potential method of detecting autophagy. The use of autophagy inhibitors including Bafilomycin A1 (Baf A1) and hydroxychloroquine (HCQ) is also a prominent method of detecting autophagy [41, 62]. Recent studies have reported that the OMM, IMM, and matrix proteins are degraded via mitophagy, whereas certain proteins regulating mitochondrial division and migration are degraded by proteasomes or ATP-dependent protease pathways. TIM23, TOM20, Hsp60, and electron transport chain (ETC)-related proteins are commonly used as mitophagy markers [63]. P62 can be recruited individually, since it is highly ubiquitinated after mitochondrial damage. P62 is involved in numerous signal transduction pathways and its function is complex. Therefore, the role of P62 in mitophagy detection is controversial [64, 65]. ...
Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation
2
2011
... A commonly used method to detect autophagy proteins is western blotting. Autophagy can be detected on the basis of the ratio of the LC3B II/I type marker protein and the extent of P62 degradation [61]. Mitophagy is accompanied by the degradation of several mitochondrial proteins; hence, it may also be evaluated by measuring their levels. Mitochondrial degradation occurs through the autophagy pathway, and cytosolic LC3 protein is converted from type I to type II via conjugation with phosphatidylethanolamine. Therefore, LC3 detection is also a potential method of detecting autophagy. The use of autophagy inhibitors including Bafilomycin A1 (Baf A1) and hydroxychloroquine (HCQ) is also a prominent method of detecting autophagy [41, 62]. Recent studies have reported that the OMM, IMM, and matrix proteins are degraded via mitophagy, whereas certain proteins regulating mitochondrial division and migration are degraded by proteasomes or ATP-dependent protease pathways. TIM23, TOM20, Hsp60, and electron transport chain (ETC)-related proteins are commonly used as mitophagy markers [63]. P62 can be recruited individually, since it is highly ubiquitinated after mitochondrial damage. P62 is involved in numerous signal transduction pathways and its function is complex. Therefore, the role of P62 in mitophagy detection is controversial [64, 65]. ...
... GFP-LC3 is often used as an autophagosome marker. Following plasmid transfection, autophagosome formation may be assessed using a fluorescence microscope. Similarly, plasmids containing fluorescent markers or specific antibodies against certain mitochondrial proteins including TOM20 and cytochrome C can be used to label mitochondria. Several commercialized mitochondrial dyes such as MitoTracker and TMRE (Tetramethylrhodamine ethyl ester) are also potentially applicable. In general, mitochondria and autophagosomes/lysosomes can be labeled separately and the colocalization of mitochondria, autophagosomes, and lysosomes may be detected via confocal laser microscopy to detect mitophagy. LC3 fused with fluorescent proteins, such as GFP and RFP, helps observe mitochondrial incorporation into autophagosomes, accompanied with the extension of the isolation membrane through time-lapse microscopic imaging of live cells. GFP fluorescence is attenuated under acidic conditions; hence, GFP-LC3 does not label autolysosomes. However, RFP is resistant to the acidic environment of lysosomes. By harnessing the distinct properties of GFP and RFP, GFP-RFP-tandem tagging LC3 is a potentially useful tool to quantify mitophagy and helps distinguish autophagosomes and autolysosomes. However, this evaluation may overlook the previously reported mitophagy mediated via an LC3-independent mechanism [66-68]. Recently, a mitochondrial probe MitoTimer was developed, which is a red fluorescent protein Ds Red "mutant" and encodes a redox sensitive mitochondrial targeted protein that switches from green to red fluorescence upon oxidation [69]. Therefore, MitoTimer is an ROS-dependent fluorophore. Furthermore, changes in the transition from green to red fluorescence are affected by temperature, oxygen, and light exposure; hence, this assay must be performed combinatorially with other methods to accurately determine the causes of changes in red or green fluorescence levels. Moreover, MitoTimer is a very useful tool to indirectly quantify mitophagy [70]. This assay quantifies several pathophysiological parameters of mitochondria, accurately assesses their health status, and plays potentially important roles in future studies [62, 71, 72]. Moreover, mitoKeima is also a fluorophore that can help quantify mitophagy. Changes in the emission wavelength of mitoKeima depend on the acidity (red color) or neutrality (green color) of the pH of the microenvironment, e.g., during the transfer from an autophagosome (green color) to a lysosome (red color). Hence, mitoKeima is a powerful pH-dependent fluorophore that is more specific for monitoring mitophagy than MitoTimer [73, 74]. ...
Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation
1
2018
... A commonly used method to detect autophagy proteins is western blotting. Autophagy can be detected on the basis of the ratio of the LC3B II/I type marker protein and the extent of P62 degradation [61]. Mitophagy is accompanied by the degradation of several mitochondrial proteins; hence, it may also be evaluated by measuring their levels. Mitochondrial degradation occurs through the autophagy pathway, and cytosolic LC3 protein is converted from type I to type II via conjugation with phosphatidylethanolamine. Therefore, LC3 detection is also a potential method of detecting autophagy. The use of autophagy inhibitors including Bafilomycin A1 (Baf A1) and hydroxychloroquine (HCQ) is also a prominent method of detecting autophagy [41, 62]. Recent studies have reported that the OMM, IMM, and matrix proteins are degraded via mitophagy, whereas certain proteins regulating mitochondrial division and migration are degraded by proteasomes or ATP-dependent protease pathways. TIM23, TOM20, Hsp60, and electron transport chain (ETC)-related proteins are commonly used as mitophagy markers [63]. P62 can be recruited individually, since it is highly ubiquitinated after mitochondrial damage. P62 is involved in numerous signal transduction pathways and its function is complex. Therefore, the role of P62 in mitophagy detection is controversial [64, 65]. ...
p62 at the crossroads of autophagy, apoptosis, and cancer
1
2009
... A commonly used method to detect autophagy proteins is western blotting. Autophagy can be detected on the basis of the ratio of the LC3B II/I type marker protein and the extent of P62 degradation [61]. Mitophagy is accompanied by the degradation of several mitochondrial proteins; hence, it may also be evaluated by measuring their levels. Mitochondrial degradation occurs through the autophagy pathway, and cytosolic LC3 protein is converted from type I to type II via conjugation with phosphatidylethanolamine. Therefore, LC3 detection is also a potential method of detecting autophagy. The use of autophagy inhibitors including Bafilomycin A1 (Baf A1) and hydroxychloroquine (HCQ) is also a prominent method of detecting autophagy [41, 62]. Recent studies have reported that the OMM, IMM, and matrix proteins are degraded via mitophagy, whereas certain proteins regulating mitochondrial division and migration are degraded by proteasomes or ATP-dependent protease pathways. TIM23, TOM20, Hsp60, and electron transport chain (ETC)-related proteins are commonly used as mitophagy markers [63]. P62 can be recruited individually, since it is highly ubiquitinated after mitochondrial damage. P62 is involved in numerous signal transduction pathways and its function is complex. Therefore, the role of P62 in mitophagy detection is controversial [64, 65]. ...
Autophagy suppresses tumorigenesis through elimination of p62
1
2009
... A commonly used method to detect autophagy proteins is western blotting. Autophagy can be detected on the basis of the ratio of the LC3B II/I type marker protein and the extent of P62 degradation [61]. Mitophagy is accompanied by the degradation of several mitochondrial proteins; hence, it may also be evaluated by measuring their levels. Mitochondrial degradation occurs through the autophagy pathway, and cytosolic LC3 protein is converted from type I to type II via conjugation with phosphatidylethanolamine. Therefore, LC3 detection is also a potential method of detecting autophagy. The use of autophagy inhibitors including Bafilomycin A1 (Baf A1) and hydroxychloroquine (HCQ) is also a prominent method of detecting autophagy [41, 62]. Recent studies have reported that the OMM, IMM, and matrix proteins are degraded via mitophagy, whereas certain proteins regulating mitochondrial division and migration are degraded by proteasomes or ATP-dependent protease pathways. TIM23, TOM20, Hsp60, and electron transport chain (ETC)-related proteins are commonly used as mitophagy markers [63]. P62 can be recruited individually, since it is highly ubiquitinated after mitochondrial damage. P62 is involved in numerous signal transduction pathways and its function is complex. Therefore, the role of P62 in mitophagy detection is controversial [64, 65]. ...
Discovery of Atg5/Atg7-independent alternative macroautophagy
1
2009
... GFP-LC3 is often used as an autophagosome marker. Following plasmid transfection, autophagosome formation may be assessed using a fluorescence microscope. Similarly, plasmids containing fluorescent markers or specific antibodies against certain mitochondrial proteins including TOM20 and cytochrome C can be used to label mitochondria. Several commercialized mitochondrial dyes such as MitoTracker and TMRE (Tetramethylrhodamine ethyl ester) are also potentially applicable. In general, mitochondria and autophagosomes/lysosomes can be labeled separately and the colocalization of mitochondria, autophagosomes, and lysosomes may be detected via confocal laser microscopy to detect mitophagy. LC3 fused with fluorescent proteins, such as GFP and RFP, helps observe mitochondrial incorporation into autophagosomes, accompanied with the extension of the isolation membrane through time-lapse microscopic imaging of live cells. GFP fluorescence is attenuated under acidic conditions; hence, GFP-LC3 does not label autolysosomes. However, RFP is resistant to the acidic environment of lysosomes. By harnessing the distinct properties of GFP and RFP, GFP-RFP-tandem tagging LC3 is a potentially useful tool to quantify mitophagy and helps distinguish autophagosomes and autolysosomes. However, this evaluation may overlook the previously reported mitophagy mediated via an LC3-independent mechanism [66-68]. Recently, a mitochondrial probe MitoTimer was developed, which is a red fluorescent protein Ds Red "mutant" and encodes a redox sensitive mitochondrial targeted protein that switches from green to red fluorescence upon oxidation [69]. Therefore, MitoTimer is an ROS-dependent fluorophore. Furthermore, changes in the transition from green to red fluorescence are affected by temperature, oxygen, and light exposure; hence, this assay must be performed combinatorially with other methods to accurately determine the causes of changes in red or green fluorescence levels. Moreover, MitoTimer is a very useful tool to indirectly quantify mitophagy [70]. This assay quantifies several pathophysiological parameters of mitochondria, accurately assesses their health status, and plays potentially important roles in future studies [62, 71, 72]. Moreover, mitoKeima is also a fluorophore that can help quantify mitophagy. Changes in the emission wavelength of mitoKeima depend on the acidity (red color) or neutrality (green color) of the pH of the microenvironment, e.g., during the transfer from an autophagosome (green color) to a lysosome (red color). Hence, mitoKeima is a powerful pH-dependent fluorophore that is more specific for monitoring mitophagy than MitoTimer [73, 74]. ...
Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation
0
2009
Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes
1
2014
... GFP-LC3 is often used as an autophagosome marker. Following plasmid transfection, autophagosome formation may be assessed using a fluorescence microscope. Similarly, plasmids containing fluorescent markers or specific antibodies against certain mitochondrial proteins including TOM20 and cytochrome C can be used to label mitochondria. Several commercialized mitochondrial dyes such as MitoTracker and TMRE (Tetramethylrhodamine ethyl ester) are also potentially applicable. In general, mitochondria and autophagosomes/lysosomes can be labeled separately and the colocalization of mitochondria, autophagosomes, and lysosomes may be detected via confocal laser microscopy to detect mitophagy. LC3 fused with fluorescent proteins, such as GFP and RFP, helps observe mitochondrial incorporation into autophagosomes, accompanied with the extension of the isolation membrane through time-lapse microscopic imaging of live cells. GFP fluorescence is attenuated under acidic conditions; hence, GFP-LC3 does not label autolysosomes. However, RFP is resistant to the acidic environment of lysosomes. By harnessing the distinct properties of GFP and RFP, GFP-RFP-tandem tagging LC3 is a potentially useful tool to quantify mitophagy and helps distinguish autophagosomes and autolysosomes. However, this evaluation may overlook the previously reported mitophagy mediated via an LC3-independent mechanism [66-68]. Recently, a mitochondrial probe MitoTimer was developed, which is a red fluorescent protein Ds Red "mutant" and encodes a redox sensitive mitochondrial targeted protein that switches from green to red fluorescence upon oxidation [69]. Therefore, MitoTimer is an ROS-dependent fluorophore. Furthermore, changes in the transition from green to red fluorescence are affected by temperature, oxygen, and light exposure; hence, this assay must be performed combinatorially with other methods to accurately determine the causes of changes in red or green fluorescence levels. Moreover, MitoTimer is a very useful tool to indirectly quantify mitophagy [70]. This assay quantifies several pathophysiological parameters of mitochondria, accurately assesses their health status, and plays potentially important roles in future studies [62, 71, 72]. Moreover, mitoKeima is also a fluorophore that can help quantify mitophagy. Changes in the emission wavelength of mitoKeima depend on the acidity (red color) or neutrality (green color) of the pH of the microenvironment, e.g., during the transfer from an autophagosome (green color) to a lysosome (red color). Hence, mitoKeima is a powerful pH-dependent fluorophore that is more specific for monitoring mitophagy than MitoTimer [73, 74]. ...
Conditional MitoTimer reporter mice for assessment of mitochondrial structure, oxidative stress, and mitophagy
1
2019
... GFP-LC3 is often used as an autophagosome marker. Following plasmid transfection, autophagosome formation may be assessed using a fluorescence microscope. Similarly, plasmids containing fluorescent markers or specific antibodies against certain mitochondrial proteins including TOM20 and cytochrome C can be used to label mitochondria. Several commercialized mitochondrial dyes such as MitoTracker and TMRE (Tetramethylrhodamine ethyl ester) are also potentially applicable. In general, mitochondria and autophagosomes/lysosomes can be labeled separately and the colocalization of mitochondria, autophagosomes, and lysosomes may be detected via confocal laser microscopy to detect mitophagy. LC3 fused with fluorescent proteins, such as GFP and RFP, helps observe mitochondrial incorporation into autophagosomes, accompanied with the extension of the isolation membrane through time-lapse microscopic imaging of live cells. GFP fluorescence is attenuated under acidic conditions; hence, GFP-LC3 does not label autolysosomes. However, RFP is resistant to the acidic environment of lysosomes. By harnessing the distinct properties of GFP and RFP, GFP-RFP-tandem tagging LC3 is a potentially useful tool to quantify mitophagy and helps distinguish autophagosomes and autolysosomes. However, this evaluation may overlook the previously reported mitophagy mediated via an LC3-independent mechanism [66-68]. Recently, a mitochondrial probe MitoTimer was developed, which is a red fluorescent protein Ds Red "mutant" and encodes a redox sensitive mitochondrial targeted protein that switches from green to red fluorescence upon oxidation [69]. Therefore, MitoTimer is an ROS-dependent fluorophore. Furthermore, changes in the transition from green to red fluorescence are affected by temperature, oxygen, and light exposure; hence, this assay must be performed combinatorially with other methods to accurately determine the causes of changes in red or green fluorescence levels. Moreover, MitoTimer is a very useful tool to indirectly quantify mitophagy [70]. This assay quantifies several pathophysiological parameters of mitochondria, accurately assesses their health status, and plays potentially important roles in future studies [62, 71, 72]. Moreover, mitoKeima is also a fluorophore that can help quantify mitophagy. Changes in the emission wavelength of mitoKeima depend on the acidity (red color) or neutrality (green color) of the pH of the microenvironment, e.g., during the transfer from an autophagosome (green color) to a lysosome (red color). Hence, mitoKeima is a powerful pH-dependent fluorophore that is more specific for monitoring mitophagy than MitoTimer [73, 74]. ...
New methods for monitoring mitochondrial biogenesis and mitophagy in vitro and in vivo
1
2017
... GFP-LC3 is often used as an autophagosome marker. Following plasmid transfection, autophagosome formation may be assessed using a fluorescence microscope. Similarly, plasmids containing fluorescent markers or specific antibodies against certain mitochondrial proteins including TOM20 and cytochrome C can be used to label mitochondria. Several commercialized mitochondrial dyes such as MitoTracker and TMRE (Tetramethylrhodamine ethyl ester) are also potentially applicable. In general, mitochondria and autophagosomes/lysosomes can be labeled separately and the colocalization of mitochondria, autophagosomes, and lysosomes may be detected via confocal laser microscopy to detect mitophagy. LC3 fused with fluorescent proteins, such as GFP and RFP, helps observe mitochondrial incorporation into autophagosomes, accompanied with the extension of the isolation membrane through time-lapse microscopic imaging of live cells. GFP fluorescence is attenuated under acidic conditions; hence, GFP-LC3 does not label autolysosomes. However, RFP is resistant to the acidic environment of lysosomes. By harnessing the distinct properties of GFP and RFP, GFP-RFP-tandem tagging LC3 is a potentially useful tool to quantify mitophagy and helps distinguish autophagosomes and autolysosomes. However, this evaluation may overlook the previously reported mitophagy mediated via an LC3-independent mechanism [66-68]. Recently, a mitochondrial probe MitoTimer was developed, which is a red fluorescent protein Ds Red "mutant" and encodes a redox sensitive mitochondrial targeted protein that switches from green to red fluorescence upon oxidation [69]. Therefore, MitoTimer is an ROS-dependent fluorophore. Furthermore, changes in the transition from green to red fluorescence are affected by temperature, oxygen, and light exposure; hence, this assay must be performed combinatorially with other methods to accurately determine the causes of changes in red or green fluorescence levels. Moreover, MitoTimer is a very useful tool to indirectly quantify mitophagy [70]. This assay quantifies several pathophysiological parameters of mitochondria, accurately assesses their health status, and plays potentially important roles in future studies [62, 71, 72]. Moreover, mitoKeima is also a fluorophore that can help quantify mitophagy. Changes in the emission wavelength of mitoKeima depend on the acidity (red color) or neutrality (green color) of the pH of the microenvironment, e.g., during the transfer from an autophagosome (green color) to a lysosome (red color). Hence, mitoKeima is a powerful pH-dependent fluorophore that is more specific for monitoring mitophagy than MitoTimer [73, 74]. ...
MitoTimer probe reveals the impact of autophagy, fusion, and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age
1
2013
... GFP-LC3 is often used as an autophagosome marker. Following plasmid transfection, autophagosome formation may be assessed using a fluorescence microscope. Similarly, plasmids containing fluorescent markers or specific antibodies against certain mitochondrial proteins including TOM20 and cytochrome C can be used to label mitochondria. Several commercialized mitochondrial dyes such as MitoTracker and TMRE (Tetramethylrhodamine ethyl ester) are also potentially applicable. In general, mitochondria and autophagosomes/lysosomes can be labeled separately and the colocalization of mitochondria, autophagosomes, and lysosomes may be detected via confocal laser microscopy to detect mitophagy. LC3 fused with fluorescent proteins, such as GFP and RFP, helps observe mitochondrial incorporation into autophagosomes, accompanied with the extension of the isolation membrane through time-lapse microscopic imaging of live cells. GFP fluorescence is attenuated under acidic conditions; hence, GFP-LC3 does not label autolysosomes. However, RFP is resistant to the acidic environment of lysosomes. By harnessing the distinct properties of GFP and RFP, GFP-RFP-tandem tagging LC3 is a potentially useful tool to quantify mitophagy and helps distinguish autophagosomes and autolysosomes. However, this evaluation may overlook the previously reported mitophagy mediated via an LC3-independent mechanism [66-68]. Recently, a mitochondrial probe MitoTimer was developed, which is a red fluorescent protein Ds Red "mutant" and encodes a redox sensitive mitochondrial targeted protein that switches from green to red fluorescence upon oxidation [69]. Therefore, MitoTimer is an ROS-dependent fluorophore. Furthermore, changes in the transition from green to red fluorescence are affected by temperature, oxygen, and light exposure; hence, this assay must be performed combinatorially with other methods to accurately determine the causes of changes in red or green fluorescence levels. Moreover, MitoTimer is a very useful tool to indirectly quantify mitophagy [70]. This assay quantifies several pathophysiological parameters of mitochondria, accurately assesses their health status, and plays potentially important roles in future studies [62, 71, 72]. Moreover, mitoKeima is also a fluorophore that can help quantify mitophagy. Changes in the emission wavelength of mitoKeima depend on the acidity (red color) or neutrality (green color) of the pH of the microenvironment, e.g., during the transfer from an autophagosome (green color) to a lysosome (red color). Hence, mitoKeima is a powerful pH-dependent fluorophore that is more specific for monitoring mitophagy than MitoTimer [73, 74]. ...
MitoTimer: a novel tool for monitoring mitochondrial turnover
1
2013
... GFP-LC3 is often used as an autophagosome marker. Following plasmid transfection, autophagosome formation may be assessed using a fluorescence microscope. Similarly, plasmids containing fluorescent markers or specific antibodies against certain mitochondrial proteins including TOM20 and cytochrome C can be used to label mitochondria. Several commercialized mitochondrial dyes such as MitoTracker and TMRE (Tetramethylrhodamine ethyl ester) are also potentially applicable. In general, mitochondria and autophagosomes/lysosomes can be labeled separately and the colocalization of mitochondria, autophagosomes, and lysosomes may be detected via confocal laser microscopy to detect mitophagy. LC3 fused with fluorescent proteins, such as GFP and RFP, helps observe mitochondrial incorporation into autophagosomes, accompanied with the extension of the isolation membrane through time-lapse microscopic imaging of live cells. GFP fluorescence is attenuated under acidic conditions; hence, GFP-LC3 does not label autolysosomes. However, RFP is resistant to the acidic environment of lysosomes. By harnessing the distinct properties of GFP and RFP, GFP-RFP-tandem tagging LC3 is a potentially useful tool to quantify mitophagy and helps distinguish autophagosomes and autolysosomes. However, this evaluation may overlook the previously reported mitophagy mediated via an LC3-independent mechanism [66-68]. Recently, a mitochondrial probe MitoTimer was developed, which is a red fluorescent protein Ds Red "mutant" and encodes a redox sensitive mitochondrial targeted protein that switches from green to red fluorescence upon oxidation [69]. Therefore, MitoTimer is an ROS-dependent fluorophore. Furthermore, changes in the transition from green to red fluorescence are affected by temperature, oxygen, and light exposure; hence, this assay must be performed combinatorially with other methods to accurately determine the causes of changes in red or green fluorescence levels. Moreover, MitoTimer is a very useful tool to indirectly quantify mitophagy [70]. This assay quantifies several pathophysiological parameters of mitochondria, accurately assesses their health status, and plays potentially important roles in future studies [62, 71, 72]. Moreover, mitoKeima is also a fluorophore that can help quantify mitophagy. Changes in the emission wavelength of mitoKeima depend on the acidity (red color) or neutrality (green color) of the pH of the microenvironment, e.g., during the transfer from an autophagosome (green color) to a lysosome (red color). Hence, mitoKeima is a powerful pH-dependent fluorophore that is more specific for monitoring mitophagy than MitoTimer [73, 74]. ...
Evaluating mitochondrial autophagy in the mouse heart
1
2016
... GFP-LC3 is often used as an autophagosome marker. Following plasmid transfection, autophagosome formation may be assessed using a fluorescence microscope. Similarly, plasmids containing fluorescent markers or specific antibodies against certain mitochondrial proteins including TOM20 and cytochrome C can be used to label mitochondria. Several commercialized mitochondrial dyes such as MitoTracker and TMRE (Tetramethylrhodamine ethyl ester) are also potentially applicable. In general, mitochondria and autophagosomes/lysosomes can be labeled separately and the colocalization of mitochondria, autophagosomes, and lysosomes may be detected via confocal laser microscopy to detect mitophagy. LC3 fused with fluorescent proteins, such as GFP and RFP, helps observe mitochondrial incorporation into autophagosomes, accompanied with the extension of the isolation membrane through time-lapse microscopic imaging of live cells. GFP fluorescence is attenuated under acidic conditions; hence, GFP-LC3 does not label autolysosomes. However, RFP is resistant to the acidic environment of lysosomes. By harnessing the distinct properties of GFP and RFP, GFP-RFP-tandem tagging LC3 is a potentially useful tool to quantify mitophagy and helps distinguish autophagosomes and autolysosomes. However, this evaluation may overlook the previously reported mitophagy mediated via an LC3-independent mechanism [66-68]. Recently, a mitochondrial probe MitoTimer was developed, which is a red fluorescent protein Ds Red "mutant" and encodes a redox sensitive mitochondrial targeted protein that switches from green to red fluorescence upon oxidation [69]. Therefore, MitoTimer is an ROS-dependent fluorophore. Furthermore, changes in the transition from green to red fluorescence are affected by temperature, oxygen, and light exposure; hence, this assay must be performed combinatorially with other methods to accurately determine the causes of changes in red or green fluorescence levels. Moreover, MitoTimer is a very useful tool to indirectly quantify mitophagy [70]. This assay quantifies several pathophysiological parameters of mitochondria, accurately assesses their health status, and plays potentially important roles in future studies [62, 71, 72]. Moreover, mitoKeima is also a fluorophore that can help quantify mitophagy. Changes in the emission wavelength of mitoKeima depend on the acidity (red color) or neutrality (green color) of the pH of the microenvironment, e.g., during the transfer from an autophagosome (green color) to a lysosome (red color). Hence, mitoKeima is a powerful pH-dependent fluorophore that is more specific for monitoring mitophagy than MitoTimer [73, 74]. ...
Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy
1
2019
... GFP-LC3 is often used as an autophagosome marker. Following plasmid transfection, autophagosome formation may be assessed using a fluorescence microscope. Similarly, plasmids containing fluorescent markers or specific antibodies against certain mitochondrial proteins including TOM20 and cytochrome C can be used to label mitochondria. Several commercialized mitochondrial dyes such as MitoTracker and TMRE (Tetramethylrhodamine ethyl ester) are also potentially applicable. In general, mitochondria and autophagosomes/lysosomes can be labeled separately and the colocalization of mitochondria, autophagosomes, and lysosomes may be detected via confocal laser microscopy to detect mitophagy. LC3 fused with fluorescent proteins, such as GFP and RFP, helps observe mitochondrial incorporation into autophagosomes, accompanied with the extension of the isolation membrane through time-lapse microscopic imaging of live cells. GFP fluorescence is attenuated under acidic conditions; hence, GFP-LC3 does not label autolysosomes. However, RFP is resistant to the acidic environment of lysosomes. By harnessing the distinct properties of GFP and RFP, GFP-RFP-tandem tagging LC3 is a potentially useful tool to quantify mitophagy and helps distinguish autophagosomes and autolysosomes. However, this evaluation may overlook the previously reported mitophagy mediated via an LC3-independent mechanism [66-68]. Recently, a mitochondrial probe MitoTimer was developed, which is a red fluorescent protein Ds Red "mutant" and encodes a redox sensitive mitochondrial targeted protein that switches from green to red fluorescence upon oxidation [69]. Therefore, MitoTimer is an ROS-dependent fluorophore. Furthermore, changes in the transition from green to red fluorescence are affected by temperature, oxygen, and light exposure; hence, this assay must be performed combinatorially with other methods to accurately determine the causes of changes in red or green fluorescence levels. Moreover, MitoTimer is a very useful tool to indirectly quantify mitophagy [70]. This assay quantifies several pathophysiological parameters of mitochondria, accurately assesses their health status, and plays potentially important roles in future studies [62, 71, 72]. Moreover, mitoKeima is also a fluorophore that can help quantify mitophagy. Changes in the emission wavelength of mitoKeima depend on the acidity (red color) or neutrality (green color) of the pH of the microenvironment, e.g., during the transfer from an autophagosome (green color) to a lysosome (red color). Hence, mitoKeima is a powerful pH-dependent fluorophore that is more specific for monitoring mitophagy than MitoTimer [73, 74]. ...
Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha
1
2006
... Mitophagy is accompanied by a reduction in mitochondrial mass; therefore, direct detection of changes in mitochondrial mass helps detect mitophagy. Mitochondria are labeled with mitochondrial dyes or corresponding antibodies, and the number of mitochondria per cell is determined via flow cytometry. RT-PCR may be used to quantify mtDNA [75-77]. These methods directly quantify mitochondrial mass. Simultaneously, mitochondrial mass is regulated via numerous regulatory mechanisms, and mitochondrial biogenesis and degradation are reportedly two of the most prominent regulatory mechanisms. Therefore, the decline in mitochondrial mass may not only be affected by mitophagy but also a process involving multiple regulatory mechanisms. Hence, determination of mitochondrial mass is an adjunct method of detecting mitophagy [78]. ...
Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane
0
2011
Labeling mitochondria with MitoTracker dyes
1
2011
... Mitophagy is accompanied by a reduction in mitochondrial mass; therefore, direct detection of changes in mitochondrial mass helps detect mitophagy. Mitochondria are labeled with mitochondrial dyes or corresponding antibodies, and the number of mitochondria per cell is determined via flow cytometry. RT-PCR may be used to quantify mtDNA [75-77]. These methods directly quantify mitochondrial mass. Simultaneously, mitochondrial mass is regulated via numerous regulatory mechanisms, and mitochondrial biogenesis and degradation are reportedly two of the most prominent regulatory mechanisms. Therefore, the decline in mitochondrial mass may not only be affected by mitophagy but also a process involving multiple regulatory mechanisms. Hence, determination of mitochondrial mass is an adjunct method of detecting mitophagy [78]. ...
Mitochondrial degradation and energy metabolism
1
2015
... Mitophagy is accompanied by a reduction in mitochondrial mass; therefore, direct detection of changes in mitochondrial mass helps detect mitophagy. Mitochondria are labeled with mitochondrial dyes or corresponding antibodies, and the number of mitochondria per cell is determined via flow cytometry. RT-PCR may be used to quantify mtDNA [75-77]. These methods directly quantify mitochondrial mass. Simultaneously, mitochondrial mass is regulated via numerous regulatory mechanisms, and mitochondrial biogenesis and degradation are reportedly two of the most prominent regulatory mechanisms. Therefore, the decline in mitochondrial mass may not only be affected by mitophagy but also a process involving multiple regulatory mechanisms. Hence, determination of mitochondrial mass is an adjunct method of detecting mitophagy [78]. ...
Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress
1
2015
... In the cardiovascular system, mitophagy exerts protective effects in response to cellular starvation. Mitophagy is activated during starvation to regulate mitochondrial quality and quantity through elimination of damaged or redundant mitochondria. Mitophagy is enhanced during cellular hypoxia. Acetylcholine reduces the damage caused by hypoxia/reoxygenation by increasing mitophagy in H9C2 cells. Violent exercise alters mitophagy levels by adaptively regulating mitochondrial function [79-81]. ...
Acute Exercise-Induced Mitochondrial Stress Triggers an Inflammatory Response in the Myocardium via NLRP3 Inflammasome Activation with Mitophagy
2
2016
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain environmental stimuli can inhibit or promote mitophagy and alter the content of the proteins related to this phenomenon. Mild hypothermia downregulated Parkin in hippocampal neurons in a cardiac arrest model, diminishing mitophagy in the neurons, protecting mitochondria, and improving neurological function after cardiac arrest [194]. Hypoxic preconditioning induced FUNDC1-dependent activation of mitophagy and decreased I/R-induced cardiac injury [96]. Myocardial mitochondrial function adapts to stress during acute exercise and manifests as significant upregulation of the mitophagy-related protein BNIP3, which stimulates mitophagy and minimizes myocardial injury [80]. Simultaneously, exercise preconditioning significantly suppresses exhaustive exercise-induced hypoxia-ischemia injuries through upregulated parkin-dependent mitophagy [195]. Acidic postconditioning-induced mitophagy renders the brain resistant to ischemic stroke through recruitment of PARK2 to mitochondria [196]. Remote ischemic post-conditioning promoted mitophagy via Parkin upregulation and inhibited oxidative stress responses, thus mitigating cerebral I/R injury in a rat model of stroke [197]. ...
Door to Intravenous Tissue Plasminogen Activator Time and Hospital Length of Stay in Acute Ischemic Stroke Patients, Georgia, 2007-2013
1
2016
... In the cardiovascular system, mitophagy exerts protective effects in response to cellular starvation. Mitophagy is activated during starvation to regulate mitochondrial quality and quantity through elimination of damaged or redundant mitochondria. Mitophagy is enhanced during cellular hypoxia. Acetylcholine reduces the damage caused by hypoxia/reoxygenation by increasing mitophagy in H9C2 cells. Violent exercise alters mitophagy levels by adaptively regulating mitochondrial function [79-81]. ...
Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction
1
2013
... During intracellular pathogenic infections, mitophagy is induced to defend against pathogens. Pathogens also induce mitophagy and disrupt mitochondrial homeostasis. For example, sepsis induces reversible cardiac mitochondrial injury, and recovery is reportedly associated with mitophagy based on morphological and biochemical evidence, suggesting that mitophagy is activated and plays an important role during cardiac recovery from sepsis [82, 83]. ...
To Explore the Protective Mechanism of PTEN-Induced Kinase 1 (PINK1)/Parkin Mitophagy-Mediated Extract of Periplaneta Americana on Lipopolysaccharide-Induced Cardiomyocyte Injury
1
2019
... During intracellular pathogenic infections, mitophagy is induced to defend against pathogens. Pathogens also induce mitophagy and disrupt mitochondrial homeostasis. For example, sepsis induces reversible cardiac mitochondrial injury, and recovery is reportedly associated with mitophagy based on morphological and biochemical evidence, suggesting that mitophagy is activated and plays an important role during cardiac recovery from sepsis [82, 83]. ...
Cardioprotection and lifespan extension by the natural polyamine spermidine
2
2016
... In normal cells, mitophagy may have a housekeeping function and renew mitochondrial components. Mitophagy participates in metabolic remodeling and is closely associated with inflammatory response factors. For example, orally administered polyaminospermine in mice enhances mitophagy, inhibits subclinical inflammation, and increases longevity. Changes in the degree of mitophagy modulate stem cell activity and chronic cardiomyocyte inflammation [19, 84]. ...
... Mitophagy alters the oxidative respiratory chain activity by regulating mitochondrial quantity and quality, thus contributing to the regulation of cell growth and development. For example, macrophage fibrosis factor (Mff)-deficient mice died of dilated cardiomyopathy at 13 weeks. Mitophagy was significantly elevated in their mutated tissues [84, 85]. ...
Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy
1
2015
... Mitophagy alters the oxidative respiratory chain activity by regulating mitochondrial quantity and quality, thus contributing to the regulation of cell growth and development. For example, macrophage fibrosis factor (Mff)-deficient mice died of dilated cardiomyopathy at 13 weeks. Mitophagy was significantly elevated in their mutated tissues [84, 85]. ...
Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart
1
2013
... Cellular aging has a prominent mitochondrial basis. Mitophagy reportedly mediates various degenerative diseases and contributes to their pathophysiology and treatment. For example, mitophagy is disrupted in the heart of aged individuals and in senescent cells and DOX-treated hearts and cells [19, 86]. ...
There is more to life than revascularization: therapeutic targeting of myocardial ischemia/reperfusion injury
1
2011
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
Temporal trends in ischemic heart disease mortality in 21 world regions, 1980 to 2010: the Global Burden of Disease 2010 study
1
2014
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS
2
2014
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
... , 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies
1
2017
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
Novel insights into the role of mitochondrial fusion and fission in cardiomyocyte apoptosis induced by ischemia/reperfusion
1
2018
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
Mitochondria and mitophagy: the yin and yang of cell death control
1
2012
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy
3
2017
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Mitophagy imbalance in cardiomyocyte ischaemia/reperfusion injury
1
2018
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
Panax Notoginseng Saponins Attenuate Myocardial Ischemia-Reperfusion Injury Through the HIF-1alpha/BNIP3 Pathway of Autophagy
1
2019
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury
5
2017
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... [96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
... Certain environmental stimuli can inhibit or promote mitophagy and alter the content of the proteins related to this phenomenon. Mild hypothermia downregulated Parkin in hippocampal neurons in a cardiac arrest model, diminishing mitophagy in the neurons, protecting mitochondria, and improving neurological function after cardiac arrest [194]. Hypoxic preconditioning induced FUNDC1-dependent activation of mitophagy and decreased I/R-induced cardiac injury [96]. Myocardial mitochondrial function adapts to stress during acute exercise and manifests as significant upregulation of the mitophagy-related protein BNIP3, which stimulates mitophagy and minimizes myocardial injury [80]. Simultaneously, exercise preconditioning significantly suppresses exhaustive exercise-induced hypoxia-ischemia injuries through upregulated parkin-dependent mitophagy [195]. Acidic postconditioning-induced mitophagy renders the brain resistant to ischemic stroke through recruitment of PARK2 to mitochondria [196]. Remote ischemic post-conditioning promoted mitophagy via Parkin upregulation and inhibited oxidative stress responses, thus mitigating cerebral I/R injury in a rat model of stroke [197]. ...
Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy
1
2007
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Mst1 promotes cardiac ischemia-reperfusion injury by inhibiting the ERK-CREB pathway and repressing FUNDC1-mediated mitophagy
1
2019
... Ischemic heart disease refers to myocardial damage caused by an imbalance between coronary blood flow and myocardial demand. Ischemic heart disease is a serious threat to human health. Ischemia causes myocardial cell injury and death [29]. Modern therapies can restore coronary circulation and achieve reperfusion, thus potentially reducing cardiac damage. Nevertheless, the recovery of blood supply has an irreversible adverse effect known as ischemia/reperfusion (I/R) injury [87, 88]. As the energy-supplying organelles in cardiomyocytes, mitochondria are major targets of I/R injury. I/R induces the loss of mitochondrial cristae, reduces mitochondrial membrane potential, and opens the mitochondrial permeability transition pore (mPTP) [89]. These effects lead to mitochondrial damage and an aggravation of the imbalance of mitophagy (excessive inhibition or promotion) [28, 89, 90]. Mitophagy protects myocardial cells during I/R injury. I/R reduces mitophagy and stimulates apoptosis in cardiomyocytes [91]. An appropriate increase in mitophagy potentially mitigates I/R injury-induced cardiomyocyte apoptosis. Simultaneously, however, mitophagy may also play a negative role in I/R injury. Inhibition of mitophagy potentially protects the myocardium against I/R injury, reduce cardiomyocyte apoptosis, improve cardiac function, and protect mitochondrial integrity[15, 92, 93]. Key mitochondrial markers PINK1 and Parkin were reportedly upregulated and enhanced Parkin transfer and activation during I/R injury [94]. I/R injury promotes BNIP3 upregulation and FUNDC1 downregulation [95, 96]. LC3 affinity for BNIP3 and FUNDC1 depends upon its phosphorylation levels. I/R injury potentially increases BNIP3 phosphorylation at S17, enhance the binding affinity of LC3, and elevate mitophagy. During I/R, phosphorylation at Y18 and S13 are increased, LC3 affinity decreased with a reduction in mitophagy [97-99]. ...
Mitochondrial dynamics in diabetic cardiomyopathy
1
2015
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage
1
2000
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Myocardial cell death in human diabetes
1
2000
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy
0
2006
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release
1
2014
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways
1
2014
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice
2
2005
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
... , 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes
0
2004
Mitochondrial respiratory impairment in streptozotocin-induced diabetic rat heart
1
1996
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes
2
2013
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
... ]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Autophagy anomalies in the diabetic myocardium
0
2011
Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice
0
2011
Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice
0
2011
Aggravated Postinfarct Heart Failure in Type 2 Diabetes Is Associated with Impaired Mitophagy and Exaggerated Inflammasome Activation
1
2017
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Diastolic dysfunction in prediabetic male rats: Role of mitochondrial oxidative stress
1
2016
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Altered regulation of the PINK1 locus: a link between type 2 diabetes and neurodegeneration?
1
2007
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Phosphatase and tensin homolog-induced putative kinase 1 and Parkin in diabetic heart: Role of mitophagy
1
2015
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
... Diabetic cardiomyopathy (DCM) is a major manifestation of organ damage in diabetic complications. DCM is an important cause of morbidity and mortality in individuals with diabetes. Mitochondrial dysfunction is a prominent contributor to diabetic cardiomyopathy [100]. Mitochondria are the primary sources of ROS and targets for oxidative stress injury. In the diabetic myocardium, damaged mitochondria produce large amounts of ROS, which exacerbates mitochondrial damage and triggers a vicious cycle of cardiomyocyte death [101-104]. In individuals with diabetic cardiomyopathy and in animal and cellular models, impaired mitochondrial structure and function and increased ROS production have been commonly observed. In vivo analyses using animals revealed that antioxidant therapy protects against diabetic cardiomyopathy [105]. However, clinical antioxidant therapy did not exert the same effect possibly because of the sustained release of ROS from damaged mitochondria [106]. Blocking of continuous ROS synthesis via mitophagy is a potential key strategy to treat diabetic cardiomyopathy [102, 106-108]. Nevertheless, the functional status and regulatory mechanisms of mitophagy in the diabetic myocardium are unknown. Mitophagy levels in a diabetic cardiomyopathy model were significantly different from those in the control. While cardiac mitophagy was inhibited in a type 1 diabetic mouse model, it was actually promoted in a type 2 diabetic mouse model [109-113]. It is yet unclear whether mitophagy protects against or induces injury in the pathogenesis of diabetic cardiomyopathy. Relative to the control, the contents of mitophagy-related proteins were significantly altered in several diabetic tissues including the heart. In male prediabetic rats, the generation of mitochondrial ROS increased and BNIP3 was downregulated in comparison with the corresponding values in the controls [14, 114]. In types 1 and 2 diabetes models, PINK1 and Parkin were downregulated [109, 115, 116]. In the vascular smooth muscle cells of diabetic mice, PINK1 and Parkin were upregulated and mitophagy was induced relative to the control [117]. ...
Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association
1
2016
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
Oxidative stress in heart failure. More than just damage
2
2003
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
... ]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
Mitochondrial Therapies in Heart Failure
0
2017
Mitochondrial dysfunction in heart failure
1
2013
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
Mitophagy and heart failure
3
2015
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
... ]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
... ]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
Xin Fu Kang oral liquid inhibits excessive myocardial mitophagy in a rat model of advanced heart failure
1
2018
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
Markers of autophagy are downregulated in failing human heart after mechanical unloading
1
2009
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
Impact of the renin-angiotensin system on cardiac energy metabolism in heart failure
1
2013
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
AMPKalpha2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation
1
2018
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
Regulating Renewable Energy: Connecting AMPKalpha2 to PINK1/Parkin-Mediated Mitophagy in the Heart
1
2018
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function
1
2011
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation
1
2008
... Heart failure (HF) is characterized by high morbidity and mortality and is the terminal stage in various heart diseases [118, 119]. Current methods of treating heart failure may mitigate or alleviate symptoms; however, the prognosis remains poor. To better determine treatment methods for heart failure, it is necessary to understand oxidative stress and chronic inflammation associated with this condition [119-121]. During heart failure, mitochondrial dysfunction is a common pathophysiological phenomenon, which has received increasing attention. Mitophagy is decreased with aging or disease [122]. Consequently, elimination of damaged mitochondria is inadequate, ROS and peroxide levels are increased. Cardiomyocyte mitochondrial proteins, lipids, and DNA undergo oxidative damage and contribute to heart failure. These findings corroborate the clinical presentation of heart failure in aged patients and those with terminal heart disease [17, 26, 123]. Biopsies of individuals with heart failure have revealed that the autophagy-specific genes beclin1 and LC3-II were downregulated after treatment [124]. Therefore, we predicted that mitophagy is associated with the occurrence and development of heart failure. Mfn2 regulates heart failure-related mitophagy by altering the mitochondrial membrane potential [125]. Subsequent experiments on heart failure models have revealed that insufficient mitophagy aggravates heart injury[122]. PINK1 is generally downregulated in individuals with heart failure, indicating a reduction in mitophagy levels. However, it is uncertain whether PINK1 downregulation is the cause or effect of heart failure [126, 127]. In vivo studies using animals confirmed the role of PINK1 in heart failure. PINK1 knockout mice were more susceptible to overload-induced heart stress and consequent heart failure than wild type mice [128]. Parkin-mediated mitophagy decreases with age [122]. Moreover, the number of dysfunctional mitochondria increases with age until it surpasses the Parkin-mediated scavenging ability of mitophagy. With aging, myocardial mitophagy becomes inadequate to maintain the normal homeostatic of mitochondrial function. In mice, Parkin deficiency has resulted in the accumulation of dysfunctional cardiomyocyte mitochondria, which elevates the risk of heart failure after myocardial infarction [28]. In mouse heart failure models induced via increased stress, Nix-silenced mice presented less myocardial fibrosis and more stable systolic function than wild type mice [129]. Thus far, myocardial mitophagy has not been reportedly increased in individuals with heart failure. Nevertheless, an increase in mitophagy leads to excessive mitochondrial clearance, and the remaining mitochondria cannot accommodate the energy demand of cardiomyocytes. This situation is extremely unfavorable for individuals with heart failure ...
Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017
1
2018
... Hypertension is a systemic disease with a high incidence and is closely associated with cardiovascular, cerebrovascular, kidney, and other diseases [130]. There is no direct evidence for the role of mitophagy in the pathophysiology of hypertension. Complications of hypertension such as atherosclerosis, myocardial I/R injury and hypertrophy, and heart failure are closely associated with cardiomyocyte and endothelial mitochondrial dysfunction [131]. Insufficient energy supply, kinetic imbalance, oxidative damage, abnormal signal transduction, and mitochondrial dysfunction caused by genetic mutations are risk factors for hypertension [132-134]. Myocardial hypertrophy associated with hypertension reportedly altered cardiomyocyte phenotype, function, and energy metabolism. These changes were reflected in the utilization of metabolic substrates and the function of the ETC, ATP synthesis, and other processes [135]. ROS regulate vascular structure and tension remodeling. An increase or retention of ROS may cause vasoconstriction, decrease vascular diastolic function, damage smooth muscle cells, promote vascular inflammation and remodeling, increase peripheral vascular resistance, and aggravate hypertension [136]. Mfn1, Mfn2, and OPA1 (optic atrophy 1) mRNA levels, which are associated with mitochondrial dynamics, were decreased in hypertensive rat heart models. Hence, mitochondrial fragmentation is increased in hypertension [137]. Mitochondrial division is a compensatory mechanism maintaining cardiac contraction in hypertension and exacerbating ventricular wall thickening [138]. Hypertension is suggested to alter mitochondrial function, energy metabolism, and mitochondrial dynamics in cardiomyocytes [131]. Clearance of mitochondrial dysfunction is closely associated with mitophagy. Therefore, elucidation of the role of mitophagy in the occurrence and development of hypertension may be important to investigate the pathophysiology of this condition and novel treatments for this condition. ...
Role of Mitochondrial Dysfunction in Hypertension and Obesity
2
2017
... Hypertension is a systemic disease with a high incidence and is closely associated with cardiovascular, cerebrovascular, kidney, and other diseases [130]. There is no direct evidence for the role of mitophagy in the pathophysiology of hypertension. Complications of hypertension such as atherosclerosis, myocardial I/R injury and hypertrophy, and heart failure are closely associated with cardiomyocyte and endothelial mitochondrial dysfunction [131]. Insufficient energy supply, kinetic imbalance, oxidative damage, abnormal signal transduction, and mitochondrial dysfunction caused by genetic mutations are risk factors for hypertension [132-134]. Myocardial hypertrophy associated with hypertension reportedly altered cardiomyocyte phenotype, function, and energy metabolism. These changes were reflected in the utilization of metabolic substrates and the function of the ETC, ATP synthesis, and other processes [135]. ROS regulate vascular structure and tension remodeling. An increase or retention of ROS may cause vasoconstriction, decrease vascular diastolic function, damage smooth muscle cells, promote vascular inflammation and remodeling, increase peripheral vascular resistance, and aggravate hypertension [136]. Mfn1, Mfn2, and OPA1 (optic atrophy 1) mRNA levels, which are associated with mitochondrial dynamics, were decreased in hypertensive rat heart models. Hence, mitochondrial fragmentation is increased in hypertension [137]. Mitochondrial division is a compensatory mechanism maintaining cardiac contraction in hypertension and exacerbating ventricular wall thickening [138]. Hypertension is suggested to alter mitochondrial function, energy metabolism, and mitochondrial dynamics in cardiomyocytes [131]. Clearance of mitochondrial dysfunction is closely associated with mitophagy. Therefore, elucidation of the role of mitophagy in the occurrence and development of hypertension may be important to investigate the pathophysiology of this condition and novel treatments for this condition. ...
... ]. Hypertension is suggested to alter mitochondrial function, energy metabolism, and mitochondrial dynamics in cardiomyocytes [131]. Clearance of mitochondrial dysfunction is closely associated with mitophagy. Therefore, elucidation of the role of mitophagy in the occurrence and development of hypertension may be important to investigate the pathophysiology of this condition and novel treatments for this condition. ...
Crosstalk between mitochondrial hyperacetylation and oxidative stress in vascular dysfunction and hypertension
1
2019
... Hypertension is a systemic disease with a high incidence and is closely associated with cardiovascular, cerebrovascular, kidney, and other diseases [130]. There is no direct evidence for the role of mitophagy in the pathophysiology of hypertension. Complications of hypertension such as atherosclerosis, myocardial I/R injury and hypertrophy, and heart failure are closely associated with cardiomyocyte and endothelial mitochondrial dysfunction [131]. Insufficient energy supply, kinetic imbalance, oxidative damage, abnormal signal transduction, and mitochondrial dysfunction caused by genetic mutations are risk factors for hypertension [132-134]. Myocardial hypertrophy associated with hypertension reportedly altered cardiomyocyte phenotype, function, and energy metabolism. These changes were reflected in the utilization of metabolic substrates and the function of the ETC, ATP synthesis, and other processes [135]. ROS regulate vascular structure and tension remodeling. An increase or retention of ROS may cause vasoconstriction, decrease vascular diastolic function, damage smooth muscle cells, promote vascular inflammation and remodeling, increase peripheral vascular resistance, and aggravate hypertension [136]. Mfn1, Mfn2, and OPA1 (optic atrophy 1) mRNA levels, which are associated with mitochondrial dynamics, were decreased in hypertensive rat heart models. Hence, mitochondrial fragmentation is increased in hypertension [137]. Mitochondrial division is a compensatory mechanism maintaining cardiac contraction in hypertension and exacerbating ventricular wall thickening [138]. Hypertension is suggested to alter mitochondrial function, energy metabolism, and mitochondrial dynamics in cardiomyocytes [131]. Clearance of mitochondrial dysfunction is closely associated with mitophagy. Therefore, elucidation of the role of mitophagy in the occurrence and development of hypertension may be important to investigate the pathophysiology of this condition and novel treatments for this condition. ...
Metabolic Alterations in Cardiopulmonary Vascular Dysfunction
1
2018
... Hypertension is a systemic disease with a high incidence and is closely associated with cardiovascular, cerebrovascular, kidney, and other diseases [130]. There is no direct evidence for the role of mitophagy in the pathophysiology of hypertension. Complications of hypertension such as atherosclerosis, myocardial I/R injury and hypertrophy, and heart failure are closely associated with cardiomyocyte and endothelial mitochondrial dysfunction [131]. Insufficient energy supply, kinetic imbalance, oxidative damage, abnormal signal transduction, and mitochondrial dysfunction caused by genetic mutations are risk factors for hypertension [132-134]. Myocardial hypertrophy associated with hypertension reportedly altered cardiomyocyte phenotype, function, and energy metabolism. These changes were reflected in the utilization of metabolic substrates and the function of the ETC, ATP synthesis, and other processes [135]. ROS regulate vascular structure and tension remodeling. An increase or retention of ROS may cause vasoconstriction, decrease vascular diastolic function, damage smooth muscle cells, promote vascular inflammation and remodeling, increase peripheral vascular resistance, and aggravate hypertension [136]. Mfn1, Mfn2, and OPA1 (optic atrophy 1) mRNA levels, which are associated with mitochondrial dynamics, were decreased in hypertensive rat heart models. Hence, mitochondrial fragmentation is increased in hypertension [137]. Mitochondrial division is a compensatory mechanism maintaining cardiac contraction in hypertension and exacerbating ventricular wall thickening [138]. Hypertension is suggested to alter mitochondrial function, energy metabolism, and mitochondrial dynamics in cardiomyocytes [131]. Clearance of mitochondrial dysfunction is closely associated with mitophagy. Therefore, elucidation of the role of mitophagy in the occurrence and development of hypertension may be important to investigate the pathophysiology of this condition and novel treatments for this condition. ...
The failing heart--an engine out of fuel
1
2007
... Hypertension is a systemic disease with a high incidence and is closely associated with cardiovascular, cerebrovascular, kidney, and other diseases [130]. There is no direct evidence for the role of mitophagy in the pathophysiology of hypertension. Complications of hypertension such as atherosclerosis, myocardial I/R injury and hypertrophy, and heart failure are closely associated with cardiomyocyte and endothelial mitochondrial dysfunction [131]. Insufficient energy supply, kinetic imbalance, oxidative damage, abnormal signal transduction, and mitochondrial dysfunction caused by genetic mutations are risk factors for hypertension [132-134]. Myocardial hypertrophy associated with hypertension reportedly altered cardiomyocyte phenotype, function, and energy metabolism. These changes were reflected in the utilization of metabolic substrates and the function of the ETC, ATP synthesis, and other processes [135]. ROS regulate vascular structure and tension remodeling. An increase or retention of ROS may cause vasoconstriction, decrease vascular diastolic function, damage smooth muscle cells, promote vascular inflammation and remodeling, increase peripheral vascular resistance, and aggravate hypertension [136]. Mfn1, Mfn2, and OPA1 (optic atrophy 1) mRNA levels, which are associated with mitochondrial dynamics, were decreased in hypertensive rat heart models. Hence, mitochondrial fragmentation is increased in hypertension [137]. Mitochondrial division is a compensatory mechanism maintaining cardiac contraction in hypertension and exacerbating ventricular wall thickening [138]. Hypertension is suggested to alter mitochondrial function, energy metabolism, and mitochondrial dynamics in cardiomyocytes [131]. Clearance of mitochondrial dysfunction is closely associated with mitophagy. Therefore, elucidation of the role of mitophagy in the occurrence and development of hypertension may be important to investigate the pathophysiology of this condition and novel treatments for this condition. ...
The Future Challenge of Reactive Oxygen Species (ROS) in Hypertension: From Bench to Bed Side
1
2017
... Hypertension is a systemic disease with a high incidence and is closely associated with cardiovascular, cerebrovascular, kidney, and other diseases [130]. There is no direct evidence for the role of mitophagy in the pathophysiology of hypertension. Complications of hypertension such as atherosclerosis, myocardial I/R injury and hypertrophy, and heart failure are closely associated with cardiomyocyte and endothelial mitochondrial dysfunction [131]. Insufficient energy supply, kinetic imbalance, oxidative damage, abnormal signal transduction, and mitochondrial dysfunction caused by genetic mutations are risk factors for hypertension [132-134]. Myocardial hypertrophy associated with hypertension reportedly altered cardiomyocyte phenotype, function, and energy metabolism. These changes were reflected in the utilization of metabolic substrates and the function of the ETC, ATP synthesis, and other processes [135]. ROS regulate vascular structure and tension remodeling. An increase or retention of ROS may cause vasoconstriction, decrease vascular diastolic function, damage smooth muscle cells, promote vascular inflammation and remodeling, increase peripheral vascular resistance, and aggravate hypertension [136]. Mfn1, Mfn2, and OPA1 (optic atrophy 1) mRNA levels, which are associated with mitochondrial dynamics, were decreased in hypertensive rat heart models. Hence, mitochondrial fragmentation is increased in hypertension [137]. Mitochondrial division is a compensatory mechanism maintaining cardiac contraction in hypertension and exacerbating ventricular wall thickening [138]. Hypertension is suggested to alter mitochondrial function, energy metabolism, and mitochondrial dynamics in cardiomyocytes [131]. Clearance of mitochondrial dysfunction is closely associated with mitophagy. Therefore, elucidation of the role of mitophagy in the occurrence and development of hypertension may be important to investigate the pathophysiology of this condition and novel treatments for this condition. ...
Down-regulation of mitofusin-2 expression in cardiac hypertrophy in vitro and in vivo
1
2007
... Hypertension is a systemic disease with a high incidence and is closely associated with cardiovascular, cerebrovascular, kidney, and other diseases [130]. There is no direct evidence for the role of mitophagy in the pathophysiology of hypertension. Complications of hypertension such as atherosclerosis, myocardial I/R injury and hypertrophy, and heart failure are closely associated with cardiomyocyte and endothelial mitochondrial dysfunction [131]. Insufficient energy supply, kinetic imbalance, oxidative damage, abnormal signal transduction, and mitochondrial dysfunction caused by genetic mutations are risk factors for hypertension [132-134]. Myocardial hypertrophy associated with hypertension reportedly altered cardiomyocyte phenotype, function, and energy metabolism. These changes were reflected in the utilization of metabolic substrates and the function of the ETC, ATP synthesis, and other processes [135]. ROS regulate vascular structure and tension remodeling. An increase or retention of ROS may cause vasoconstriction, decrease vascular diastolic function, damage smooth muscle cells, promote vascular inflammation and remodeling, increase peripheral vascular resistance, and aggravate hypertension [136]. Mfn1, Mfn2, and OPA1 (optic atrophy 1) mRNA levels, which are associated with mitochondrial dynamics, were decreased in hypertensive rat heart models. Hence, mitochondrial fragmentation is increased in hypertension [137]. Mitochondrial division is a compensatory mechanism maintaining cardiac contraction in hypertension and exacerbating ventricular wall thickening [138]. Hypertension is suggested to alter mitochondrial function, energy metabolism, and mitochondrial dynamics in cardiomyocytes [131]. Clearance of mitochondrial dysfunction is closely associated with mitophagy. Therefore, elucidation of the role of mitophagy in the occurrence and development of hypertension may be important to investigate the pathophysiology of this condition and novel treatments for this condition. ...
A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy
1
2010
... Hypertension is a systemic disease with a high incidence and is closely associated with cardiovascular, cerebrovascular, kidney, and other diseases [130]. There is no direct evidence for the role of mitophagy in the pathophysiology of hypertension. Complications of hypertension such as atherosclerosis, myocardial I/R injury and hypertrophy, and heart failure are closely associated with cardiomyocyte and endothelial mitochondrial dysfunction [131]. Insufficient energy supply, kinetic imbalance, oxidative damage, abnormal signal transduction, and mitochondrial dysfunction caused by genetic mutations are risk factors for hypertension [132-134]. Myocardial hypertrophy associated with hypertension reportedly altered cardiomyocyte phenotype, function, and energy metabolism. These changes were reflected in the utilization of metabolic substrates and the function of the ETC, ATP synthesis, and other processes [135]. ROS regulate vascular structure and tension remodeling. An increase or retention of ROS may cause vasoconstriction, decrease vascular diastolic function, damage smooth muscle cells, promote vascular inflammation and remodeling, increase peripheral vascular resistance, and aggravate hypertension [136]. Mfn1, Mfn2, and OPA1 (optic atrophy 1) mRNA levels, which are associated with mitochondrial dynamics, were decreased in hypertensive rat heart models. Hence, mitochondrial fragmentation is increased in hypertension [137]. Mitochondrial division is a compensatory mechanism maintaining cardiac contraction in hypertension and exacerbating ventricular wall thickening [138]. Hypertension is suggested to alter mitochondrial function, energy metabolism, and mitochondrial dynamics in cardiomyocytes [131]. Clearance of mitochondrial dysfunction is closely associated with mitophagy. Therefore, elucidation of the role of mitophagy in the occurrence and development of hypertension may be important to investigate the pathophysiology of this condition and novel treatments for this condition. ...
New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis
1
2017
... Atherosclerosis (AS) is a chronic disease characterized by lipid accumulation, vascular smooth muscle cell proliferation, apoptosis, and local inflammation [139]. The pathogenesis of atherosclerosis is complex and includes abnormal lipid metabolism, inflammation, endothelial injury, oxidative stress, and other mechanisms [140, 141]. In the pathophysiology of atherosclerosis, plaque tissue stability determines disease occurrence and progression [142]. In plaque macrophages, vascular smooth muscle cells, and endothelial cells, mitophagy clears the damaged mitochondria, stabilizes the mitochondrial function in the damaged cells, reduces cell damage, maintains plaque integrity, and prevents disease progression caused by plaque tissue rupture [143]. Oxidized low-density lipoprotein (ox-LDL) has been widely used to generate atherosclerosis models because it is an atherosclerotic agent. In vascular smooth muscle cells, ox-LDL disrupts mitochondrial structure, increases ROS levels, and induces mitochondrial dysfunction [144]. Furthermore, Ox-LDL activates mitophagy, which eliminates nonfunctional and damaged mitochondria. After the addition of mitophagy inhibitors, mitophagy and its related pathway proteins are significantly decreased, and cell apoptosis is significantly increased [145]. Mitophagy protects against atherosclerotic stress-induced apoptosis in human vascular smooth muscle cells (VSMCs) [146-148]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques. ...
Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration
1
2008
... Atherosclerosis (AS) is a chronic disease characterized by lipid accumulation, vascular smooth muscle cell proliferation, apoptosis, and local inflammation [139]. The pathogenesis of atherosclerosis is complex and includes abnormal lipid metabolism, inflammation, endothelial injury, oxidative stress, and other mechanisms [140, 141]. In the pathophysiology of atherosclerosis, plaque tissue stability determines disease occurrence and progression [142]. In plaque macrophages, vascular smooth muscle cells, and endothelial cells, mitophagy clears the damaged mitochondria, stabilizes the mitochondrial function in the damaged cells, reduces cell damage, maintains plaque integrity, and prevents disease progression caused by plaque tissue rupture [143]. Oxidized low-density lipoprotein (ox-LDL) has been widely used to generate atherosclerosis models because it is an atherosclerotic agent. In vascular smooth muscle cells, ox-LDL disrupts mitochondrial structure, increases ROS levels, and induces mitochondrial dysfunction [144]. Furthermore, Ox-LDL activates mitophagy, which eliminates nonfunctional and damaged mitochondria. After the addition of mitophagy inhibitors, mitophagy and its related pathway proteins are significantly decreased, and cell apoptosis is significantly increased [145]. Mitophagy protects against atherosclerotic stress-induced apoptosis in human vascular smooth muscle cells (VSMCs) [146-148]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques. ...
The immune system in atherosclerosis
1
2011
... Atherosclerosis (AS) is a chronic disease characterized by lipid accumulation, vascular smooth muscle cell proliferation, apoptosis, and local inflammation [139]. The pathogenesis of atherosclerosis is complex and includes abnormal lipid metabolism, inflammation, endothelial injury, oxidative stress, and other mechanisms [140, 141]. In the pathophysiology of atherosclerosis, plaque tissue stability determines disease occurrence and progression [142]. In plaque macrophages, vascular smooth muscle cells, and endothelial cells, mitophagy clears the damaged mitochondria, stabilizes the mitochondrial function in the damaged cells, reduces cell damage, maintains plaque integrity, and prevents disease progression caused by plaque tissue rupture [143]. Oxidized low-density lipoprotein (ox-LDL) has been widely used to generate atherosclerosis models because it is an atherosclerotic agent. In vascular smooth muscle cells, ox-LDL disrupts mitochondrial structure, increases ROS levels, and induces mitochondrial dysfunction [144]. Furthermore, Ox-LDL activates mitophagy, which eliminates nonfunctional and damaged mitochondria. After the addition of mitophagy inhibitors, mitophagy and its related pathway proteins are significantly decreased, and cell apoptosis is significantly increased [145]. Mitophagy protects against atherosclerotic stress-induced apoptosis in human vascular smooth muscle cells (VSMCs) [146-148]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques. ...
Plaque Calcification During Atherosclerosis Progression and Regression
1
2018
... Atherosclerosis (AS) is a chronic disease characterized by lipid accumulation, vascular smooth muscle cell proliferation, apoptosis, and local inflammation [139]. The pathogenesis of atherosclerosis is complex and includes abnormal lipid metabolism, inflammation, endothelial injury, oxidative stress, and other mechanisms [140, 141]. In the pathophysiology of atherosclerosis, plaque tissue stability determines disease occurrence and progression [142]. In plaque macrophages, vascular smooth muscle cells, and endothelial cells, mitophagy clears the damaged mitochondria, stabilizes the mitochondrial function in the damaged cells, reduces cell damage, maintains plaque integrity, and prevents disease progression caused by plaque tissue rupture [143]. Oxidized low-density lipoprotein (ox-LDL) has been widely used to generate atherosclerosis models because it is an atherosclerotic agent. In vascular smooth muscle cells, ox-LDL disrupts mitochondrial structure, increases ROS levels, and induces mitochondrial dysfunction [144]. Furthermore, Ox-LDL activates mitophagy, which eliminates nonfunctional and damaged mitochondria. After the addition of mitophagy inhibitors, mitophagy and its related pathway proteins are significantly decreased, and cell apoptosis is significantly increased [145]. Mitophagy protects against atherosclerotic stress-induced apoptosis in human vascular smooth muscle cells (VSMCs) [146-148]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques. ...
Defective Autophagy in Atherosclerosis: To Die or to Senesce?
1
2018
... Atherosclerosis (AS) is a chronic disease characterized by lipid accumulation, vascular smooth muscle cell proliferation, apoptosis, and local inflammation [139]. The pathogenesis of atherosclerosis is complex and includes abnormal lipid metabolism, inflammation, endothelial injury, oxidative stress, and other mechanisms [140, 141]. In the pathophysiology of atherosclerosis, plaque tissue stability determines disease occurrence and progression [142]. In plaque macrophages, vascular smooth muscle cells, and endothelial cells, mitophagy clears the damaged mitochondria, stabilizes the mitochondrial function in the damaged cells, reduces cell damage, maintains plaque integrity, and prevents disease progression caused by plaque tissue rupture [143]. Oxidized low-density lipoprotein (ox-LDL) has been widely used to generate atherosclerosis models because it is an atherosclerotic agent. In vascular smooth muscle cells, ox-LDL disrupts mitochondrial structure, increases ROS levels, and induces mitochondrial dysfunction [144]. Furthermore, Ox-LDL activates mitophagy, which eliminates nonfunctional and damaged mitochondria. After the addition of mitophagy inhibitors, mitophagy and its related pathway proteins are significantly decreased, and cell apoptosis is significantly increased [145]. Mitophagy protects against atherosclerotic stress-induced apoptosis in human vascular smooth muscle cells (VSMCs) [146-148]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques. ...
Oxidative Stress in Atherosclerosis
1
2017
... Atherosclerosis (AS) is a chronic disease characterized by lipid accumulation, vascular smooth muscle cell proliferation, apoptosis, and local inflammation [139]. The pathogenesis of atherosclerosis is complex and includes abnormal lipid metabolism, inflammation, endothelial injury, oxidative stress, and other mechanisms [140, 141]. In the pathophysiology of atherosclerosis, plaque tissue stability determines disease occurrence and progression [142]. In plaque macrophages, vascular smooth muscle cells, and endothelial cells, mitophagy clears the damaged mitochondria, stabilizes the mitochondrial function in the damaged cells, reduces cell damage, maintains plaque integrity, and prevents disease progression caused by plaque tissue rupture [143]. Oxidized low-density lipoprotein (ox-LDL) has been widely used to generate atherosclerosis models because it is an atherosclerotic agent. In vascular smooth muscle cells, ox-LDL disrupts mitochondrial structure, increases ROS levels, and induces mitochondrial dysfunction [144]. Furthermore, Ox-LDL activates mitophagy, which eliminates nonfunctional and damaged mitochondria. After the addition of mitophagy inhibitors, mitophagy and its related pathway proteins are significantly decreased, and cell apoptosis is significantly increased [145]. Mitophagy protects against atherosclerotic stress-induced apoptosis in human vascular smooth muscle cells (VSMCs) [146-148]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques. ...
NR4A1 contributes to high-fat associated endothelial dysfunction by promoting CaMKII-Parkin-mitophagy pathways
3
2018
... Atherosclerosis (AS) is a chronic disease characterized by lipid accumulation, vascular smooth muscle cell proliferation, apoptosis, and local inflammation [139]. The pathogenesis of atherosclerosis is complex and includes abnormal lipid metabolism, inflammation, endothelial injury, oxidative stress, and other mechanisms [140, 141]. In the pathophysiology of atherosclerosis, plaque tissue stability determines disease occurrence and progression [142]. In plaque macrophages, vascular smooth muscle cells, and endothelial cells, mitophagy clears the damaged mitochondria, stabilizes the mitochondrial function in the damaged cells, reduces cell damage, maintains plaque integrity, and prevents disease progression caused by plaque tissue rupture [143]. Oxidized low-density lipoprotein (ox-LDL) has been widely used to generate atherosclerosis models because it is an atherosclerotic agent. In vascular smooth muscle cells, ox-LDL disrupts mitochondrial structure, increases ROS levels, and induces mitochondrial dysfunction [144]. Furthermore, Ox-LDL activates mitophagy, which eliminates nonfunctional and damaged mitochondria. After the addition of mitophagy inhibitors, mitophagy and its related pathway proteins are significantly decreased, and cell apoptosis is significantly increased [145]. Mitophagy protects against atherosclerotic stress-induced apoptosis in human vascular smooth muscle cells (VSMCs) [146-148]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques. ...
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Several signaling pathways regulate molecules that control mitophagy. In a mouse atherosclerosis model established with ox-LDL, NR4A1 was significantly upregulated and controlled Parkin activation through posttranscriptional CaMKII modification. Thus, it activated Parkin-mediated mitophagy. Excessive mitophagy reduced mitochondrial quality which, in turn, caused energy shortage and mitochondrial dysfunction [145]. Furthermore, blockade of the MAPK-ERK-CREB signaling pathway upregulated NR4A1 and repressed Mfn2-mediated mitophagy, thus expanding the area of cerebral infarction and increasing neuronal apoptosis [181]. In cardiomyocytes, damaged mitochondria are sequestered in rab5-positive early endosomes via the ESCRT mechanism, which depends on Parkin, and the loss of rab5 function increases the susceptibility of embryonic fibroblasts and cardiomyocytes to cell death [182]. In a mouse ischemic model, activation of the p53-TIGAR axis downregulated BNIP3, inhibited mitophagy, caused the accumulation of damaged mitochondria, and weakened the cardioprotective effect [183]. The JNK signaling pathway is a key regulator of the FOXO3a transcription factor, which promotes BNIP3 expression in heart failure models. JNK influences the degree of mitophagy by regulating the level of BNIP3[184]. ...
Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids
4
2016
... Atherosclerosis (AS) is a chronic disease characterized by lipid accumulation, vascular smooth muscle cell proliferation, apoptosis, and local inflammation [139]. The pathogenesis of atherosclerosis is complex and includes abnormal lipid metabolism, inflammation, endothelial injury, oxidative stress, and other mechanisms [140, 141]. In the pathophysiology of atherosclerosis, plaque tissue stability determines disease occurrence and progression [142]. In plaque macrophages, vascular smooth muscle cells, and endothelial cells, mitophagy clears the damaged mitochondria, stabilizes the mitochondrial function in the damaged cells, reduces cell damage, maintains plaque integrity, and prevents disease progression caused by plaque tissue rupture [143]. Oxidized low-density lipoprotein (ox-LDL) has been widely used to generate atherosclerosis models because it is an atherosclerotic agent. In vascular smooth muscle cells, ox-LDL disrupts mitochondrial structure, increases ROS levels, and induces mitochondrial dysfunction [144]. Furthermore, Ox-LDL activates mitophagy, which eliminates nonfunctional and damaged mitochondria. After the addition of mitophagy inhibitors, mitophagy and its related pathway proteins are significantly decreased, and cell apoptosis is significantly increased [145]. Mitophagy protects against atherosclerotic stress-induced apoptosis in human vascular smooth muscle cells (VSMCs) [146-148]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques. ...
... ]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques. ...
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Impaired mitochondrial respiration in human carotid plaque atherosclerosis: A potential role for Pink1 in vascular smooth muscle cell energetics
3
2018
... Atherosclerosis (AS) is a chronic disease characterized by lipid accumulation, vascular smooth muscle cell proliferation, apoptosis, and local inflammation [139]. The pathogenesis of atherosclerosis is complex and includes abnormal lipid metabolism, inflammation, endothelial injury, oxidative stress, and other mechanisms [140, 141]. In the pathophysiology of atherosclerosis, plaque tissue stability determines disease occurrence and progression [142]. In plaque macrophages, vascular smooth muscle cells, and endothelial cells, mitophagy clears the damaged mitochondria, stabilizes the mitochondrial function in the damaged cells, reduces cell damage, maintains plaque integrity, and prevents disease progression caused by plaque tissue rupture [143]. Oxidized low-density lipoprotein (ox-LDL) has been widely used to generate atherosclerosis models because it is an atherosclerotic agent. In vascular smooth muscle cells, ox-LDL disrupts mitochondrial structure, increases ROS levels, and induces mitochondrial dysfunction [144]. Furthermore, Ox-LDL activates mitophagy, which eliminates nonfunctional and damaged mitochondria. After the addition of mitophagy inhibitors, mitophagy and its related pathway proteins are significantly decreased, and cell apoptosis is significantly increased [145]. Mitophagy protects against atherosclerotic stress-induced apoptosis in human vascular smooth muscle cells (VSMCs) [146-148]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques. ...
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Autophagy and Mitophagy in Cardiovascular Disease
1
2017
... Atherosclerosis (AS) is a chronic disease characterized by lipid accumulation, vascular smooth muscle cell proliferation, apoptosis, and local inflammation [139]. The pathogenesis of atherosclerosis is complex and includes abnormal lipid metabolism, inflammation, endothelial injury, oxidative stress, and other mechanisms [140, 141]. In the pathophysiology of atherosclerosis, plaque tissue stability determines disease occurrence and progression [142]. In plaque macrophages, vascular smooth muscle cells, and endothelial cells, mitophagy clears the damaged mitochondria, stabilizes the mitochondrial function in the damaged cells, reduces cell damage, maintains plaque integrity, and prevents disease progression caused by plaque tissue rupture [143]. Oxidized low-density lipoprotein (ox-LDL) has been widely used to generate atherosclerosis models because it is an atherosclerotic agent. In vascular smooth muscle cells, ox-LDL disrupts mitochondrial structure, increases ROS levels, and induces mitochondrial dysfunction [144]. Furthermore, Ox-LDL activates mitophagy, which eliminates nonfunctional and damaged mitochondria. After the addition of mitophagy inhibitors, mitophagy and its related pathway proteins are significantly decreased, and cell apoptosis is significantly increased [145]. Mitophagy protects against atherosclerotic stress-induced apoptosis in human vascular smooth muscle cells (VSMCs) [146-148]. PINK1/Parkin was reportedly upregulated in atherosclerotic disease models than in normal tissues. PINK1 overexpression enhances the protective effect of mitophagy on VSMCs, whereas PINK1 gene knockout counteracts it [146, 147]. Hence, mitophagy is a potential target for the stabilization of atherosclerotic plaques. ...
Atrial fibrillation and microRNAs
1
2014
... Arrhythmia is a prominent aspect of CVD. It may occur individually or in association with other cardiovascular disease. In a healthy heart, coordinated electrical propagation supports cardiac function; upon failure of such a cardiac action potential, cardiac arrhythmia occurs [149]. Although there is no direct evidence of the role of mitophagy in arrhythmia, extensive evidence indicates that mitochondria play a functional role in arrhythmia. Mitochondria are involved in arrhythmia owing to their ability to produce ATP and ROS [150]. Mitochondrial dysfunction reduces ATP production, affects cardiac electrical conduction, thus altering sarcolemmal K+ fluxes via ATP-sensitive potassium channels [151]. However, excessive mitochondrial ROS production can introduce heterogeneity in cardiac action potentials [152]. Alterations in ATP and ROS levels are based on a vicious cycle: during mitochondrial dysfunction, the mitochondrial membrane potential reduced, thus decreasing ATP and increasing ROS levels; thereafter, further mitochondrial dysfunction is induced, with the mitochondrial membrane potential reduced, with a concomitant reduction in ATP generation. This vicious cycle causes electrophysiological alterations, thereby causing arrhythmia [150, 153]. Proper mitophagy can eliminate damaged mitochondria, maintain mitochondrial homeostasis in cells, and prevent damage to hazardous substances, thus further disrupting cellular phenomena. During arrhythmia, changes in ATP and ROS levels caused by mitochondrial damage play an important role in mitophagy. Therefore, we can boldly speculate that mitophagy eliminates damaged mitochondria and regulates ROS and ATP levels to a certain extent, thereby inhibiting or decelerating the occurrence of arrhythmia. ...
Mitochondria and arrhythmias
2
2014
... Arrhythmia is a prominent aspect of CVD. It may occur individually or in association with other cardiovascular disease. In a healthy heart, coordinated electrical propagation supports cardiac function; upon failure of such a cardiac action potential, cardiac arrhythmia occurs [149]. Although there is no direct evidence of the role of mitophagy in arrhythmia, extensive evidence indicates that mitochondria play a functional role in arrhythmia. Mitochondria are involved in arrhythmia owing to their ability to produce ATP and ROS [150]. Mitochondrial dysfunction reduces ATP production, affects cardiac electrical conduction, thus altering sarcolemmal K+ fluxes via ATP-sensitive potassium channels [151]. However, excessive mitochondrial ROS production can introduce heterogeneity in cardiac action potentials [152]. Alterations in ATP and ROS levels are based on a vicious cycle: during mitochondrial dysfunction, the mitochondrial membrane potential reduced, thus decreasing ATP and increasing ROS levels; thereafter, further mitochondrial dysfunction is induced, with the mitochondrial membrane potential reduced, with a concomitant reduction in ATP generation. This vicious cycle causes electrophysiological alterations, thereby causing arrhythmia [150, 153]. Proper mitophagy can eliminate damaged mitochondria, maintain mitochondrial homeostasis in cells, and prevent damage to hazardous substances, thus further disrupting cellular phenomena. During arrhythmia, changes in ATP and ROS levels caused by mitochondrial damage play an important role in mitophagy. Therefore, we can boldly speculate that mitophagy eliminates damaged mitochondria and regulates ROS and ATP levels to a certain extent, thereby inhibiting or decelerating the occurrence of arrhythmia. ...
... ]. Alterations in ATP and ROS levels are based on a vicious cycle: during mitochondrial dysfunction, the mitochondrial membrane potential reduced, thus decreasing ATP and increasing ROS levels; thereafter, further mitochondrial dysfunction is induced, with the mitochondrial membrane potential reduced, with a concomitant reduction in ATP generation. This vicious cycle causes electrophysiological alterations, thereby causing arrhythmia [150, 153]. Proper mitophagy can eliminate damaged mitochondria, maintain mitochondrial homeostasis in cells, and prevent damage to hazardous substances, thus further disrupting cellular phenomena. During arrhythmia, changes in ATP and ROS levels caused by mitochondrial damage play an important role in mitophagy. Therefore, we can boldly speculate that mitophagy eliminates damaged mitochondria and regulates ROS and ATP levels to a certain extent, thereby inhibiting or decelerating the occurrence of arrhythmia. ...
ATP-sensitive potassium channel blocker HMR 1883 reduces mortality and ischemia-associated electrocardiographic changes in pigs with coronary occlusion
1
1999
... Arrhythmia is a prominent aspect of CVD. It may occur individually or in association with other cardiovascular disease. In a healthy heart, coordinated electrical propagation supports cardiac function; upon failure of such a cardiac action potential, cardiac arrhythmia occurs [149]. Although there is no direct evidence of the role of mitophagy in arrhythmia, extensive evidence indicates that mitochondria play a functional role in arrhythmia. Mitochondria are involved in arrhythmia owing to their ability to produce ATP and ROS [150]. Mitochondrial dysfunction reduces ATP production, affects cardiac electrical conduction, thus altering sarcolemmal K+ fluxes via ATP-sensitive potassium channels [151]. However, excessive mitochondrial ROS production can introduce heterogeneity in cardiac action potentials [152]. Alterations in ATP and ROS levels are based on a vicious cycle: during mitochondrial dysfunction, the mitochondrial membrane potential reduced, thus decreasing ATP and increasing ROS levels; thereafter, further mitochondrial dysfunction is induced, with the mitochondrial membrane potential reduced, with a concomitant reduction in ATP generation. This vicious cycle causes electrophysiological alterations, thereby causing arrhythmia [150, 153]. Proper mitophagy can eliminate damaged mitochondria, maintain mitochondrial homeostasis in cells, and prevent damage to hazardous substances, thus further disrupting cellular phenomena. During arrhythmia, changes in ATP and ROS levels caused by mitochondrial damage play an important role in mitophagy. Therefore, we can boldly speculate that mitophagy eliminates damaged mitochondria and regulates ROS and ATP levels to a certain extent, thereby inhibiting or decelerating the occurrence of arrhythmia. ...
Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation
1
2006
... Arrhythmia is a prominent aspect of CVD. It may occur individually or in association with other cardiovascular disease. In a healthy heart, coordinated electrical propagation supports cardiac function; upon failure of such a cardiac action potential, cardiac arrhythmia occurs [149]. Although there is no direct evidence of the role of mitophagy in arrhythmia, extensive evidence indicates that mitochondria play a functional role in arrhythmia. Mitochondria are involved in arrhythmia owing to their ability to produce ATP and ROS [150]. Mitochondrial dysfunction reduces ATP production, affects cardiac electrical conduction, thus altering sarcolemmal K+ fluxes via ATP-sensitive potassium channels [151]. However, excessive mitochondrial ROS production can introduce heterogeneity in cardiac action potentials [152]. Alterations in ATP and ROS levels are based on a vicious cycle: during mitochondrial dysfunction, the mitochondrial membrane potential reduced, thus decreasing ATP and increasing ROS levels; thereafter, further mitochondrial dysfunction is induced, with the mitochondrial membrane potential reduced, with a concomitant reduction in ATP generation. This vicious cycle causes electrophysiological alterations, thereby causing arrhythmia [150, 153]. Proper mitophagy can eliminate damaged mitochondria, maintain mitochondrial homeostasis in cells, and prevent damage to hazardous substances, thus further disrupting cellular phenomena. During arrhythmia, changes in ATP and ROS levels caused by mitochondrial damage play an important role in mitophagy. Therefore, we can boldly speculate that mitophagy eliminates damaged mitochondria and regulates ROS and ATP levels to a certain extent, thereby inhibiting or decelerating the occurrence of arrhythmia. ...
Functional Role of Mitochondria in Arrhythmogenesis
1
2017
... Arrhythmia is a prominent aspect of CVD. It may occur individually or in association with other cardiovascular disease. In a healthy heart, coordinated electrical propagation supports cardiac function; upon failure of such a cardiac action potential, cardiac arrhythmia occurs [149]. Although there is no direct evidence of the role of mitophagy in arrhythmia, extensive evidence indicates that mitochondria play a functional role in arrhythmia. Mitochondria are involved in arrhythmia owing to their ability to produce ATP and ROS [150]. Mitochondrial dysfunction reduces ATP production, affects cardiac electrical conduction, thus altering sarcolemmal K+ fluxes via ATP-sensitive potassium channels [151]. However, excessive mitochondrial ROS production can introduce heterogeneity in cardiac action potentials [152]. Alterations in ATP and ROS levels are based on a vicious cycle: during mitochondrial dysfunction, the mitochondrial membrane potential reduced, thus decreasing ATP and increasing ROS levels; thereafter, further mitochondrial dysfunction is induced, with the mitochondrial membrane potential reduced, with a concomitant reduction in ATP generation. This vicious cycle causes electrophysiological alterations, thereby causing arrhythmia [150, 153]. Proper mitophagy can eliminate damaged mitochondria, maintain mitochondrial homeostasis in cells, and prevent damage to hazardous substances, thus further disrupting cellular phenomena. During arrhythmia, changes in ATP and ROS levels caused by mitochondrial damage play an important role in mitophagy. Therefore, we can boldly speculate that mitophagy eliminates damaged mitochondria and regulates ROS and ATP levels to a certain extent, thereby inhibiting or decelerating the occurrence of arrhythmia. ...
Mechanisms of brain injury after intracerebral haemorrhage
1
2006
... Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163]. ...
Advances in the management of intracerebral hemorrhage
1
2013
... Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163]. ...
Emerging medical and surgical management strategies in the evaluation and treatment of intracerebral hemorrhage
1
2005
... Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163]. ...
Chemical Conditioning as an Approach to Ischemic Stroke Tolerance: Mitochondria as the Target
1
2016
... Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163]. ...
Stroke neuroprotection: targeting mitochondria
1
2013
... Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163]. ...
Perihematomal mitochondrial dysfunction after intracerebral hemorrhage
1
2006
... Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163]. ...
BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke
1
2014
... Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163]. ...
BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2
3
2017
... Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163]. ...
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Mitophagy is activated in brain damage induced by cerebral ischemia and reperfusion via the PINK1/Parkin/p62 signalling pathway
1
2018
... Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163]. ...
Reactive nitrogen species as therapeutic targets for autophagy: implication for ischemic stroke
1
2017
... Stroke is an acute cerebrovascular disease, which can be categorized as ischemic stroke and hemorrhagic stroke in accordance with its pathogenesis. Ischemic stroke is caused by atherosclerotic plaque disruption and spontaneous intracranial hemorrhage and is often caused by hypertension [154, 155]. Current studies on stroke have revealed significant advancements; however, current treatments still have risks [156]. In ischemic stroke, mitochondrial aggregation events that cause cell death and tissue infarction primarily result from a lack of nutrients and oxygen and reduction in ATP, and during the reperfusion phase, extensive cellular damage are caused by a mitochondrial Ca2+ overload, mPTP opening, and excessive production of ROS [157, 158]. In hemorrhagic stroke, mitochondrial dysfunction decreases mitochondrial respiratory function, and ischemia decreases oxygen metabolism in hematoma [159]. Hence, the regulation of mitochondrial homeostasis is an apparently promising therapy for stroke, and mitophagy serves as an important underlying mechanism. However, mitophagy is a double-edged sword in stroke; there is no consensus regarding whether mitophagy plays a protective or damaging role in stroke. In in vivo and in vitro models of stroke, BNIP3 and Nix are reportedly upregulated, leading to delayed neuronal death [160]; however, subsequent studies reported that Nix-mediated mitophagy protects against ischemic stroke [161]. PINK1/Parkin-mediated mitophagy is activated in brain damage induced by ischemia stroke; however, the underlying function is unclear [162]. Although the function of mitophagy is still unclear in stroke, it is generally accepted that the degree of mitophagy is the determinant during stroke, and physiological levels can be beneficial, while excessive or inadequate levels could be deleterious [30, 163]. ...
Overview of microRNA biology
1
2015
... MicroRNAs are small, single-stranded, noncoding RNA molecules which inhibit the translation of target mRNAs or induce their degradation [164]. MiR-410 is reportedly significantly upregulated in an I/R injury model of human adult cardiomyocytes (HACMs), which presented decreased mitochondrial function and loss of mitophagy. MiR-410 overexpression reportedly decreased cell viability, ATP generation, mitochondrial membrane potential, and mitophagy. In contrast, miR-410 downregulation had the opposite effect. MiR-410 directly targeted HSPB1 and regulated its activity and mitophagy [165]. MiR-137 is reportedly significantly upregulated under hypoxia, wherein it downregulates Nix and FUNDC1 and impairs mitophagy [166]. In lipid toxicity and diabetic mouse models, miR-133a was reportedly downregulated and Nix was upregulated. MiR-133a upregulation inhibited Nix translation, regulated mitochondrial function, and stabilized mitochondrial membrane potential [167]. ...
MicroRNA-410 is involved in mitophagy after cardiac ischemia/reperfusion injury by targeting high-mobility group box 1 protein
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... MicroRNAs are small, single-stranded, noncoding RNA molecules which inhibit the translation of target mRNAs or induce their degradation [164]. MiR-410 is reportedly significantly upregulated in an I/R injury model of human adult cardiomyocytes (HACMs), which presented decreased mitochondrial function and loss of mitophagy. MiR-410 overexpression reportedly decreased cell viability, ATP generation, mitochondrial membrane potential, and mitophagy. In contrast, miR-410 downregulation had the opposite effect. MiR-410 directly targeted HSPB1 and regulated its activity and mitophagy [165]. MiR-137 is reportedly significantly upregulated under hypoxia, wherein it downregulates Nix and FUNDC1 and impairs mitophagy [166]. In lipid toxicity and diabetic mouse models, miR-133a was reportedly downregulated and Nix was upregulated. MiR-133a upregulation inhibited Nix translation, regulated mitochondrial function, and stabilized mitochondrial membrane potential [167]. ...
MicroRNA-137 is a novel hypoxia-responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX
2
2014
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... MicroRNAs are small, single-stranded, noncoding RNA molecules which inhibit the translation of target mRNAs or induce their degradation [164]. MiR-410 is reportedly significantly upregulated in an I/R injury model of human adult cardiomyocytes (HACMs), which presented decreased mitochondrial function and loss of mitophagy. MiR-410 overexpression reportedly decreased cell viability, ATP generation, mitochondrial membrane potential, and mitophagy. In contrast, miR-410 downregulation had the opposite effect. MiR-410 directly targeted HSPB1 and regulated its activity and mitophagy [165]. MiR-137 is reportedly significantly upregulated under hypoxia, wherein it downregulates Nix and FUNDC1 and impairs mitophagy [166]. In lipid toxicity and diabetic mouse models, miR-133a was reportedly downregulated and Nix was upregulated. MiR-133a upregulation inhibited Nix translation, regulated mitochondrial function, and stabilized mitochondrial membrane potential [167]. ...
A conserved MADS-box phosphorylation motif regulates differentiation and mitochondrial function in skeletal, cardiac, and smooth muscle cells
2
2015
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... MicroRNAs are small, single-stranded, noncoding RNA molecules which inhibit the translation of target mRNAs or induce their degradation [164]. MiR-410 is reportedly significantly upregulated in an I/R injury model of human adult cardiomyocytes (HACMs), which presented decreased mitochondrial function and loss of mitophagy. MiR-410 overexpression reportedly decreased cell viability, ATP generation, mitochondrial membrane potential, and mitophagy. In contrast, miR-410 downregulation had the opposite effect. MiR-410 directly targeted HSPB1 and regulated its activity and mitophagy [165]. MiR-137 is reportedly significantly upregulated under hypoxia, wherein it downregulates Nix and FUNDC1 and impairs mitophagy [166]. In lipid toxicity and diabetic mouse models, miR-133a was reportedly downregulated and Nix was upregulated. MiR-133a upregulation inhibited Nix translation, regulated mitochondrial function, and stabilized mitochondrial membrane potential [167]. ...
Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis
2
2017
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Melatonin activates Parkin translocation and rescues the impaired mitophagy activity of diabetic cardiomyopathy through Mst1 inhibition
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Mitophagy is required for acute cardioprotection by simvastatin
2
2014
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Liraglutide repairs the infarcted heart: The role of the SIRT1/Parkin/mitophagy pathway
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Zinc prevents mitochondrial superoxide generation by inducing mitophagy in the setting of hypoxia/reoxygenation in cardiac cells
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Protective Effect of Sevoflurane Postconditioning against Cardiac Ischemia/Reperfusion Injury via Ameliorating Mitochondrial Impairment, Oxidative Stress and Rescuing Autophagic Clearance
2
2015
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Restoring Pharmacologic Preconditioning in the Aging Heart: Role of Mitophagy/Autophagy
2
2017
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Bnip3 Binds and Activates p300: Possible Role in Cardiac Transcription and Myocyte Morphology
2
2015
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes
2
2017
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy
2
2014
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Inhibition of Peroxynitrite-Induced Mitophagy Activation Attenuates Cerebral Ischemia-Reperfusion Injury
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
Naringin Attenuates Cerebral Ischemia-Reperfusion Injury Through Inhibiting Peroxynitrite-Mediated Mitophagy Activation
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain drugs and chemicals regulate mitophagy. Melatonin prevents the opening of the mPTP and the activation of PINK1/Parkin in the microcirculating endothelial cells of an I/R mouse model. Melatonin prevents mitophagy-mediated cell death and protects cardiac microvessels from I/R injury; the underlying mechanism may be attributed to the inhibitory effects of melatonin on mitochondrial fission VDAC1 HK2 mPTP mitophagy axis via activation of AMPKα [168]. Furthermore, melatonin restores mitophagy in diabetic cardiomyopathy probably via Parkin translocation and Mst1 repression [169]. Subsequent studies reported that melatonin induced mitophagy through the Sirt3/FOXO3a/Parkin signaling pathway, attenuated NLRP3 (NLR family pyrin domain containing 3) inflammasome activation, and prevented the progression of atherosclerosis [170]. Simvastatin suppresses mTOR signaling, triggers mitochondrial translocation of Parkin and p62/SQSTM1, activates mitophagy, and reduces the infarct area in mouse and HL-1 cell models of myocardial infarction. Coenzyme Q may influence the anti-ischemic effect of statins by interfering with mitophagy [171]. Liraglutide upregulated SIRT1 and Parkin, activated mitophagy, reduced cellular oxidative stress, balanced the redox reaction, and maintained mitochondrial homeostasis. These effects promoted myocardial repair after myocardial infarction [172]. In both healthy and I/R rat cardiomyocytes, zinc (Zn) upregulated PINK1 and Beclin1, activated mitophagy, inhibited the generation of superoxide mitochondria, reduced the loss of mitochondrial membrane potential during reperfusion, prevented mitochondrial oxidative stress, and protected heart damage [173]. Sevoflurane posttreatment downregulated Parkin in a rat I/R model, inhibited mitophagy, maintained ATP, reversed mitochondrial damage, and protected the heart [174]. Pretreatment with the antioxidant TEMPOL upregulated PINK1 and Parkin, restored mitophagy in the aged myocardium upon isoproterenol (ISO) treatment, and promoted cardiac recovery in aging animals [175]. BNIP3 binding activated acetyltransferase p300 and increased histone acetylation and transcription factor GATA4 levels. Curcumin inhibited these phenomena and reduced mitophagy levels [176]. Erythorbic acid (EA) reduced mitochondrial injury and necrotic cell death of cardiac myocytes and alleviated oxidative damage and cardiac dysfunction during anthracycline therapy by functionally abrogating Bnip3 activity, thus inhibiting BNIP3-induced mitophagy [177]. Tunicamycin and thapsigargin-induced ER stress protects against ischemic stroke injury, which is probably involved in mitophagy induction [178]. Peroxynitrite-induced PINK1/Parkin-mediated mitophagy activation through drp1 recruitment to damaged mitochondria aggravates cerebral I/R injury during stroke [179]. A natural antioxidant naringin potentially inhibits peroxynitrite-mediated mitophagy activation by inhibiting the translocation of Parkin to the mitochondria and attenuating ischemic stroke injury [180]. ...
NR4A1 Promotes Cerebral Ischemia Reperfusion Injury by Repressing Mfn2-Mediated Mitophagy and Inactivating the MAPK-ERK-CREB Signaling Pathway
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Several signaling pathways regulate molecules that control mitophagy. In a mouse atherosclerosis model established with ox-LDL, NR4A1 was significantly upregulated and controlled Parkin activation through posttranscriptional CaMKII modification. Thus, it activated Parkin-mediated mitophagy. Excessive mitophagy reduced mitochondrial quality which, in turn, caused energy shortage and mitochondrial dysfunction [145]. Furthermore, blockade of the MAPK-ERK-CREB signaling pathway upregulated NR4A1 and repressed Mfn2-mediated mitophagy, thus expanding the area of cerebral infarction and increasing neuronal apoptosis [181]. In cardiomyocytes, damaged mitochondria are sequestered in rab5-positive early endosomes via the ESCRT mechanism, which depends on Parkin, and the loss of rab5 function increases the susceptibility of embryonic fibroblasts and cardiomyocytes to cell death [182]. In a mouse ischemic model, activation of the p53-TIGAR axis downregulated BNIP3, inhibited mitophagy, caused the accumulation of damaged mitochondria, and weakened the cardioprotective effect [183]. The JNK signaling pathway is a key regulator of the FOXO3a transcription factor, which promotes BNIP3 expression in heart failure models. JNK influences the degree of mitophagy by regulating the level of BNIP3[184]. ...
A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance
2
2017
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Several signaling pathways regulate molecules that control mitophagy. In a mouse atherosclerosis model established with ox-LDL, NR4A1 was significantly upregulated and controlled Parkin activation through posttranscriptional CaMKII modification. Thus, it activated Parkin-mediated mitophagy. Excessive mitophagy reduced mitochondrial quality which, in turn, caused energy shortage and mitochondrial dysfunction [145]. Furthermore, blockade of the MAPK-ERK-CREB signaling pathway upregulated NR4A1 and repressed Mfn2-mediated mitophagy, thus expanding the area of cerebral infarction and increasing neuronal apoptosis [181]. In cardiomyocytes, damaged mitochondria are sequestered in rab5-positive early endosomes via the ESCRT mechanism, which depends on Parkin, and the loss of rab5 function increases the susceptibility of embryonic fibroblasts and cardiomyocytes to cell death [182]. In a mouse ischemic model, activation of the p53-TIGAR axis downregulated BNIP3, inhibited mitophagy, caused the accumulation of damaged mitochondria, and weakened the cardioprotective effect [183]. The JNK signaling pathway is a key regulator of the FOXO3a transcription factor, which promotes BNIP3 expression in heart failure models. JNK influences the degree of mitophagy by regulating the level of BNIP3[184]. ...
p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia
2
2012
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Several signaling pathways regulate molecules that control mitophagy. In a mouse atherosclerosis model established with ox-LDL, NR4A1 was significantly upregulated and controlled Parkin activation through posttranscriptional CaMKII modification. Thus, it activated Parkin-mediated mitophagy. Excessive mitophagy reduced mitochondrial quality which, in turn, caused energy shortage and mitochondrial dysfunction [145]. Furthermore, blockade of the MAPK-ERK-CREB signaling pathway upregulated NR4A1 and repressed Mfn2-mediated mitophagy, thus expanding the area of cerebral infarction and increasing neuronal apoptosis [181]. In cardiomyocytes, damaged mitochondria are sequestered in rab5-positive early endosomes via the ESCRT mechanism, which depends on Parkin, and the loss of rab5 function increases the susceptibility of embryonic fibroblasts and cardiomyocytes to cell death [182]. In a mouse ischemic model, activation of the p53-TIGAR axis downregulated BNIP3, inhibited mitophagy, caused the accumulation of damaged mitochondria, and weakened the cardioprotective effect [183]. The JNK signaling pathway is a key regulator of the FOXO3a transcription factor, which promotes BNIP3 expression in heart failure models. JNK influences the degree of mitophagy by regulating the level of BNIP3[184]. ...
JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure
2
2012
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Several signaling pathways regulate molecules that control mitophagy. In a mouse atherosclerosis model established with ox-LDL, NR4A1 was significantly upregulated and controlled Parkin activation through posttranscriptional CaMKII modification. Thus, it activated Parkin-mediated mitophagy. Excessive mitophagy reduced mitochondrial quality which, in turn, caused energy shortage and mitochondrial dysfunction [145]. Furthermore, blockade of the MAPK-ERK-CREB signaling pathway upregulated NR4A1 and repressed Mfn2-mediated mitophagy, thus expanding the area of cerebral infarction and increasing neuronal apoptosis [181]. In cardiomyocytes, damaged mitochondria are sequestered in rab5-positive early endosomes via the ESCRT mechanism, which depends on Parkin, and the loss of rab5 function increases the susceptibility of embryonic fibroblasts and cardiomyocytes to cell death [182]. In a mouse ischemic model, activation of the p53-TIGAR axis downregulated BNIP3, inhibited mitophagy, caused the accumulation of damaged mitochondria, and weakened the cardioprotective effect [183]. The JNK signaling pathway is a key regulator of the FOXO3a transcription factor, which promotes BNIP3 expression in heart failure models. JNK influences the degree of mitophagy by regulating the level of BNIP3[184]. ...
Signal transducer and activator of transcription-1 localizes to the mitochondria and modulates mitophagy
2
2013
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Aldehyde Dehydrogenase 2 Has Cardioprotective Effects on Myocardial Ischaemia/Reperfusion Injury via Suppressing Mitophagy
2
2016
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy
2
2017
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Mst1 inhibits Sirt3 expression and contributes to diabetic cardiomyopathy through inhibiting Parkin-dependent mitophagy
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Evidence for the Role of BAG3 in Mitochondrial Quality Control in Cardiomyocytes
2
2017
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
PINK1/Parkin-mediated mitophagy promotes apelin-13-induced vascular smooth muscle cell proliferation by AMPKalpha and exacerbates atherosclerotic lesions
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Involvement of PINK1/Parkin-mediated mitophagy in AGE-induced cardiomyocyte aging
2
2017
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Akt2 ablation prolongs life span and improves myocardial contractile function with adaptive cardiac remodeling: role of Sirt1-mediated autophagy regulation
2
2017
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
BRG1 and BRM SWI/SNF ATPases redundantly maintain cardiomyocyte homeostasis by regulating cardiomyocyte mitophagy and mitochondrial dynamics in vivo
2
2016
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain receptor antagonists, inhibitors of upstream molecules, and gene knockouts directly or indirectly regulate mitophagy. STAT1 is not only localized in the mitochondria but also serves as an LC3b binding partner; hence, STAT1 decreases mitophagy and promotes cell death in the myocardial I/R response [185]. Silencing of PINK1 and Parkin reduced mitophagy in atherosclerosis and enhanced apoptosis in VSMCs via ox-LDL. In contrast, PINK1 and Parkin overexpression inhibited cell death [146, 147]. G protein-coupled estrogen receptor 1 (GPER) agonist increases GPER activity, reduces Parkin translocation from the cytosol to the mitochondria, and downregulates PINK1 protein expression, inhibiting the PINK1/Parkin pathway and mitophagy, protecting mitochondrial structural integrity and function, and protecting the heart after I/R injury [93]. Both in I/R rats and hypoxia/reoxygenation H9C2 cells, ALDH2 activation suppressed phosphatase and PINK1/Parkin expression, preventing 4-hydroxynonenal, ROS, and mitochondrial superoxide accumulation, and regulating mitophagy, thus protecting the heart against I/R injury [186]. In a mouse model of diabetic cardiomyopathy, Sirt3 overexpression activated deacetylation of Foxo3A and expression of Parkin, upregulated Parkin-dependent mitophagy, inhibited mitochondrial damage and apoptosis in cardiomyocytes, and played an important role in the occurrence and development of diabetic cardiomyopathy [187]. Mst1 knockout reportedly significantly upregulated Parkin, enhanced mitochondrial translocation, and protected the myocardium of diabetic mice. These effects may be associated with Sirt3 downregulation via Mst1 [188]. BAG3 functions upstream of Parkin, and BAG3 knock-down reduced PINK1/Parkin-mediated mitophagy and impaired the clearance of defective mitochondria, thus increasing levels of toxicity within the cells and subsequent cell death in the context of heart failure [189]. In AS, apelin-13 increases mitophagy by activating the PINK1/Parkin pathway, and F13A is an apelin-13 antagonist in PINK1 and Parkin-dependent mitophagy, which blocks the effect of apelin-13 in the progression of atherosclerosis [190]. Both CsA and PINK1 knockout significantly downregulated PINK1 and Parkin proteins in senescent cardiomyocytes, thus reducing the level of mitophagy and blocking cardiomyocyte senescence [191]. Akt2 knockout prevented cardiac aging by upregulating Foxo1-related BNIP3, PINK1, and Parkin and by maintaining mitochondrial integrity [192]. Nix knockout impaired mitophagy and aggravated ischemic stroke in mice, which can be rescued via Nix overexpression [161]. DUSP1 overexpression inhibited BNIP3 activation, thereby inactivating the JNK pathway and alleviating mitophagy. This mechanism improved the survival of myocardial tissue after I/R[98]. After SWI/SNF was deleted in Brg1/Brm double-mutant mice, BNIP3 was upregulated and mitophagy was increased. Consequently, small and fragmented mitochondria were formed. This reaction corresponded with changes occurring during heart failure[193]. In the I/R model of FUNDC1 knockout mice, mitophagy was inhibited and cardiac injury was aggravated [96]. ...
Mild hypothermia alleviates excessive autophagy and mitophagy in a rat model of asphyxial cardiac arrest
2
2014
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain environmental stimuli can inhibit or promote mitophagy and alter the content of the proteins related to this phenomenon. Mild hypothermia downregulated Parkin in hippocampal neurons in a cardiac arrest model, diminishing mitophagy in the neurons, protecting mitochondria, and improving neurological function after cardiac arrest [194]. Hypoxic preconditioning induced FUNDC1-dependent activation of mitophagy and decreased I/R-induced cardiac injury [96]. Myocardial mitochondrial function adapts to stress during acute exercise and manifests as significant upregulation of the mitophagy-related protein BNIP3, which stimulates mitophagy and minimizes myocardial injury [80]. Simultaneously, exercise preconditioning significantly suppresses exhaustive exercise-induced hypoxia-ischemia injuries through upregulated parkin-dependent mitophagy [195]. Acidic postconditioning-induced mitophagy renders the brain resistant to ischemic stroke through recruitment of PARK2 to mitochondria [196]. Remote ischemic post-conditioning promoted mitophagy via Parkin upregulation and inhibited oxidative stress responses, thus mitigating cerebral I/R injury in a rat model of stroke [197]. ...
Parkin Mediates Mitophagy to Participate in Cardioprotection Induced by Late Exercise Preconditioning but Bnip3 Does Not. J
2
2018
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain environmental stimuli can inhibit or promote mitophagy and alter the content of the proteins related to this phenomenon. Mild hypothermia downregulated Parkin in hippocampal neurons in a cardiac arrest model, diminishing mitophagy in the neurons, protecting mitochondria, and improving neurological function after cardiac arrest [194]. Hypoxic preconditioning induced FUNDC1-dependent activation of mitophagy and decreased I/R-induced cardiac injury [96]. Myocardial mitochondrial function adapts to stress during acute exercise and manifests as significant upregulation of the mitophagy-related protein BNIP3, which stimulates mitophagy and minimizes myocardial injury [80]. Simultaneously, exercise preconditioning significantly suppresses exhaustive exercise-induced hypoxia-ischemia injuries through upregulated parkin-dependent mitophagy [195]. Acidic postconditioning-induced mitophagy renders the brain resistant to ischemic stroke through recruitment of PARK2 to mitochondria [196]. Remote ischemic post-conditioning promoted mitophagy via Parkin upregulation and inhibited oxidative stress responses, thus mitigating cerebral I/R injury in a rat model of stroke [197]. ...
PARK2-dependent mitophagy induced by acidic postconditioning protects against focal cerebral ischemia and extends the reperfusion window
2
2017
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain environmental stimuli can inhibit or promote mitophagy and alter the content of the proteins related to this phenomenon. Mild hypothermia downregulated Parkin in hippocampal neurons in a cardiac arrest model, diminishing mitophagy in the neurons, protecting mitochondria, and improving neurological function after cardiac arrest [194]. Hypoxic preconditioning induced FUNDC1-dependent activation of mitophagy and decreased I/R-induced cardiac injury [96]. Myocardial mitochondrial function adapts to stress during acute exercise and manifests as significant upregulation of the mitophagy-related protein BNIP3, which stimulates mitophagy and minimizes myocardial injury [80]. Simultaneously, exercise preconditioning significantly suppresses exhaustive exercise-induced hypoxia-ischemia injuries through upregulated parkin-dependent mitophagy [195]. Acidic postconditioning-induced mitophagy renders the brain resistant to ischemic stroke through recruitment of PARK2 to mitochondria [196]. Remote ischemic post-conditioning promoted mitophagy via Parkin upregulation and inhibited oxidative stress responses, thus mitigating cerebral I/R injury in a rat model of stroke [197]. ...
Role of mitophagy regulated by Parkin/DJ-1 in remote ischemic postconditioning-induced mitigation of focal cerebral ischemia-reperfusion
2
2015
... Therapeutic application of mitophagy.
Diseases
Representatives
Mechanisms
Effects
References
MicroRNAs
I/R
MiR-410
Mitophagy-
damage
[165]
I/R
MiR-137
BNIP3/FUNDC1-
-
[166]
Cardiac Lipotoxicity, DCM
MiR-133a
Nix-
protection
[167]
Clinical drugs and chemical reagents
I/R
Melatonin
PINK1/Parkin-
protection
[168]
DCM
Melatonin
PINK1/Parkin+
protection
[169]
AS
Melatonin
PINK1/Parkin+
protection
[170]
I/R
Simvastatin
Parkin/P62+
protection
[171]
I/R
Liraglutide
Parkin+
protection
[172]
I/R
Zine
PINK1+
protection
[173]
I/R
Sevoflurane postconditioning
Parkin-
protection
[174]
I/R
TEMPOL preconditioning
PINK1/Parkin+
protection
[175]
HF
Curcumin
BNIP3-
protection
[176]
Cardiotoxicity
Ellagic acid
BNIP3-
protection
[177]
Stroke
Tunicamycin and thapsigargin
Mitophagy+
protection
[178]
Stroke
Peroxynitrite
PINK1/Parkin+
damage
[179]
Stroke
Naringin
Parkin-
protection
[180]
Signal pathways
AS
NR4A1/CaMKII activation
Parkin+
damage
[145]
Stroke
MAPK-ERK-CREB blockade
Mfn2-
damage
[181]
I/R
Rab5 endosomal pathway activation
Parkin+
protection
[182].
I/R
P53/TIGAR activation
BNIP3-
damage
[183]
HF
JNK/FOXO3a activation
BNIP3+
damage
[184].
Activators/inhibitors, genes knock in/out
I/R
STAT1 activation
Mitophagy-
damage
[185]
AS
PINK1/Parkin knockout
PINK1/Parkin-
damage
[146, 147]
I/R
GPER activation
PINK1/Parkin-
protection
[93]
I/R
ALDH2 activation
PINK1/Parkin-
protection
[186]
DCM
Sirt3 overexpression
Parkin+
protection
[187]
DCM
Mst1 knockout
Parkin+
protection
[188]
HF
BAG3 knockdown
Parkin-
damage
[189]
AS
F13A
PINK1/Parkin-
protection
[190]
HF
CsA
PINK1/Parkin-
protection
[191]
HF
Akt2 knockout
BNIP3/PINK1/Parkin+
protection
[192]
Stroke
Nix knockout
Nix-
damage
[161]
I/R
DUSP1 activation
BNIP3-
protection
[98]
HF
SWI/SNF deletion
BNIP3+
damage
[193]
I/R
FUNDC1 knockout
FUNDC1-
damage
[96]
Environmental stimuli
I/R
Mild hypothermia
Parkin-
protection
[194]
I/R
Hypoxic preconditioning
FUNDC1+
protection
[96]
Myocardial inflammatory
Acute exercise
BNIP3+
protection
[80]
I/R
Exercise preconditioning
Parkin+
protection
[195]
Stroke
Acidic postconditioning
Mitophagy+
protection
[196]
Stroke
Remote ischemic post conditioning
Parkin+
protection
[197]
4. Therapeutic application of mitophagy
Mitophagy plays an important role in CVD pathogenesis and is, therefore, a potential therapeutic target. CVD can be treated, or its progress can be delayed by promoting or inhibiting mitophagy, thus maintaining the functional stability of mitochondria and reducing cell damage in the disease state (Table 1). ...
... Certain environmental stimuli can inhibit or promote mitophagy and alter the content of the proteins related to this phenomenon. Mild hypothermia downregulated Parkin in hippocampal neurons in a cardiac arrest model, diminishing mitophagy in the neurons, protecting mitochondria, and improving neurological function after cardiac arrest [194]. Hypoxic preconditioning induced FUNDC1-dependent activation of mitophagy and decreased I/R-induced cardiac injury [96]. Myocardial mitochondrial function adapts to stress during acute exercise and manifests as significant upregulation of the mitophagy-related protein BNIP3, which stimulates mitophagy and minimizes myocardial injury [80]. Simultaneously, exercise preconditioning significantly suppresses exhaustive exercise-induced hypoxia-ischemia injuries through upregulated parkin-dependent mitophagy [195]. Acidic postconditioning-induced mitophagy renders the brain resistant to ischemic stroke through recruitment of PARK2 to mitochondria [196]. Remote ischemic post-conditioning promoted mitophagy via Parkin upregulation and inhibited oxidative stress responses, thus mitigating cerebral I/R injury in a rat model of stroke [197]. ...
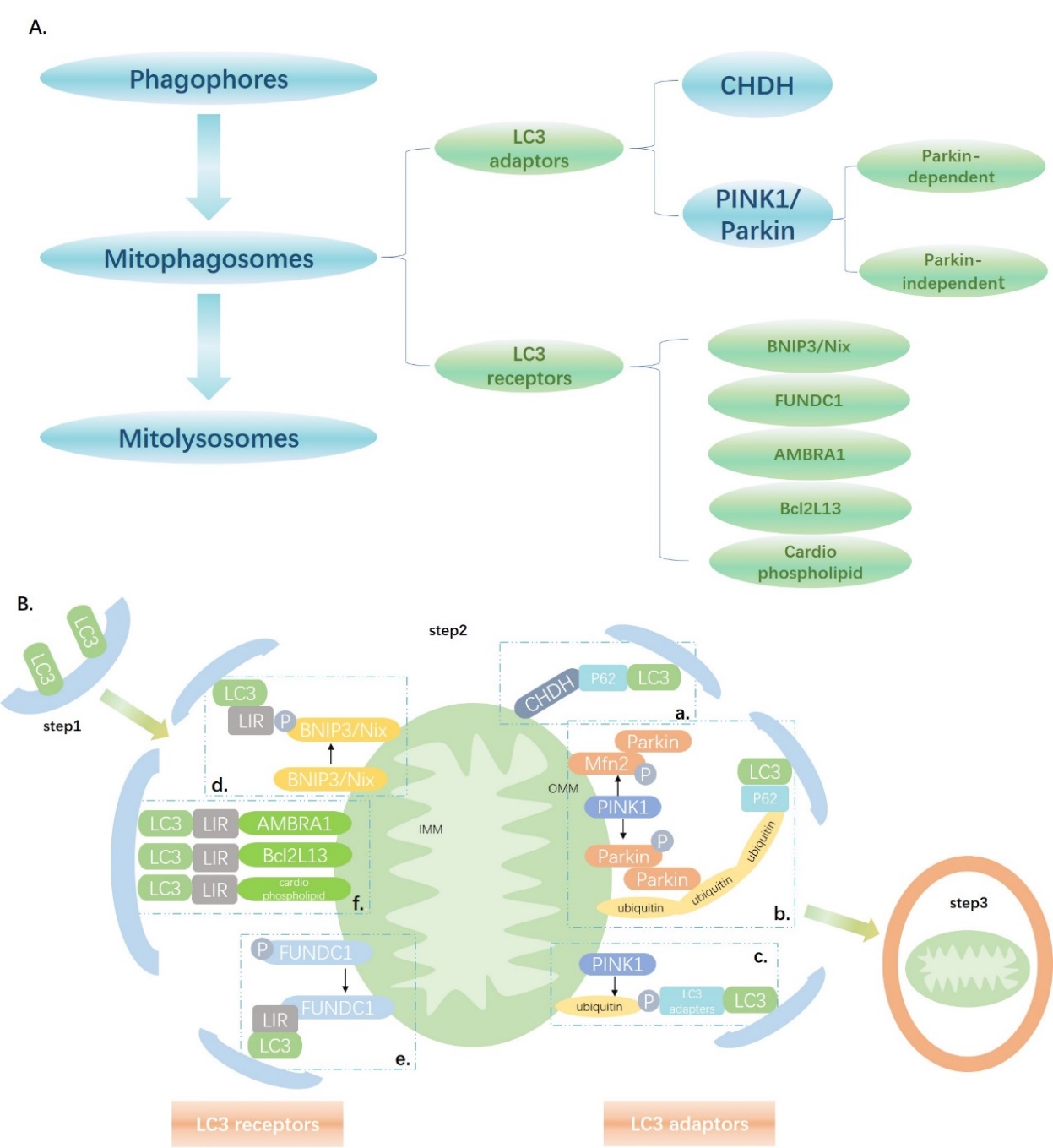
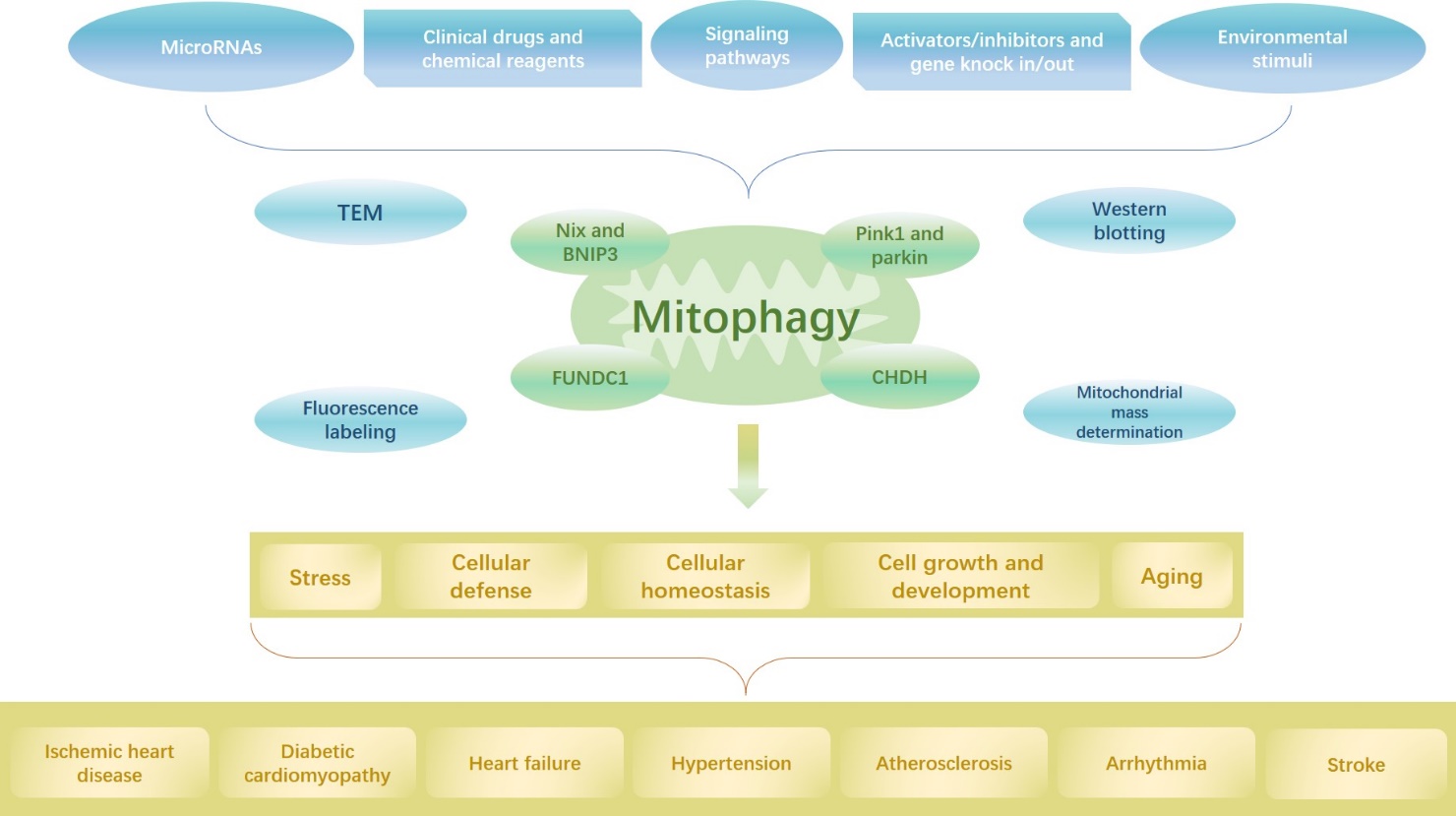
{kind=link}
{kind=link}
{kind=link}
{kind=link}



